- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 34(3); 2010 > Article
-
ReviewInflammation and Insulin Resistance: An Old Story with New Ideas
- Jason K. Kim
-
Korean Diabetes Journal 2010;34(3):137-145.
DOI: https://doi.org/10.4093/kdj.2010.34.3.137
Published online: June 30, 2010
- 3,139 Views
- 40 Download
- 9 Crossref
Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, Massachusetts, USA.
- Corresponding author: Jason K. Kim. University of Massachusetts Medical School, 381 Plantation Street, Suite 200, Worcester, MA 01605, USA. jason.kim@umassmed.edu
Copyright © 2010 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- JNK1 REGULATES ADIPOSE INFLAMMATION IN OBESITY
- PROTECTIVE ROLE OF JNK1 IN HEPATIC STEATOSIS AND INSULIN RESISTANCE
- JNK1 MEDIATES DIET-INDUCED INSULIN RESISTANCE IN SKELETAL MUSCLE
- NERVOUS SYSTEM JNK1 REGULATES ENERGY BALANCE VIA HYPOTHALAMIC-PITUITARY-THYROID AXIS
- MOLECULAR SCAFFOLD KSR2 REGULATES ENERGY BALANCE AND GLUCOSE METABOLISM
- ENDOPLASMIC RETICULUM CHAPERONE GRP78 REGULATES ENERGY BALANCE AND INSULIN RESISTANCE
- FUTURE QUESTIONS
- CONCLUSION
- REFERENCES
ABSTRACT
- Years before insulin was discovered, anti-inflammatory sodium salicylate was used to treat diabetes in 1901. Intriguingly for many years that followed, diabetes was viewed as a disorder of glucose metabolism, and then it was described as a disease of dysregulated lipid metabolism. The diabetes research focused on the causal relationship between obesity and insulin resistance, a major characteristic of type 2 diabetes. It is only within the past 20 years when the notion of inflammation as a cause of insulin resistance began to surface. In obesity, inflammation develops when macrophages infiltrate adipose tissue and stimulate adipocyte secretion of inflammatory cytokines, that in turn affect energy balance, glucose and lipid metabolism, leading to insulin resistance. This report reviews recent discoveries of stress kinase signaling involving molecular scaffolds and endoplasmic reticulum chaperones that regulate energy balance and glucose homeostasis. As we advance from a conceptual understanding to molecular discoveries, a century-old story of inflammation and insulin resistance is re-born with new ideas.
- Insulin resistance is a major characteristic of type 2 diabetes, which affects more than 250 million people worldwide [1]. Since insulin resistance is as an early and requisite event in the development of type 2 diabetes, understanding the cause of insulin resistance and identifying therapeutic intervention to treat insulin resistance have a global significance [2]. It is well established that obesity is a major cause of insulin resistance, and weight loss is known to improve insulin sensitivity in diabetic subjects [3]. However, the underlying mechanism by which obesity causes insulin resistance remains unclear. A number of interesting hypotheses and pathways have recently emerged that individually or collectively can explain the important link between obesity and insulin resistance [4,5].
- One of the most exciting topics in recent diabetes research is the role of inflammation in type 2 diabetes. In 1993, Hotamisligil et al. [6] reported the role of adipose expression tumor necrosis factor-α (TNF-α) in obesity-linked insulin resistance. This was immediately followed by a landmark discovery of obese (ob) gene by Zhang et al. in 1994 that led to a frantic search and identification of a host of adipocyte-derived hormones, such as leptin and adiponectin, that potently regulate glucose homeostasis [7,8]. The birth of "adipocentric" view of diabetes has taken place. Despite the early discovery of TNF-α as a novel inflammatory cytokine to regulate glucose metabolism, the role of inflammation in diabetes was not established until a series of clinical observations that supported laboratory findings. In 1997, Pickup et al. [9] reported a significant association between interleukin-6 (IL-6) and type 2 diabetes. This was further supported by Kern et al. [10] who described a role of adipose-derived TNF-α and IL-6 in human obesity and insulin resistance.
- At present, there is little doubt that adipose tissue is a master regulator of inflammation, and inflammation mediates insulin resistance in obesity [11]. Interestingly, this notion is not new, and a careful review of historical studies finds that Williamson [12] reported the beneficial effects of anti-inflammatory sodium salicylate on the treatment of diabetes in 1901. This review will describe recent discoveries on the underlying mechanism by which inflammation causes insulin resistance and therefore attempt to advance our understanding on a century-old notion.
INTRODUCTION
- The c-Jun NH2-terminal kinase 1 (JNK1) signaling pathway is involved in the pathogenesis of obesity, insulin resistance, and type 2 diabetes [13]. In obese state induced by chronic high-fat diet (HFD) or genetic manipulation, JNK1 is activated and mediates downstream signaling events that target glucose metabolism [14]. JNK1 has been shown to promote a serine phosphorylation of insulin receptor substrate 1 (IRS-1), and this event inhibits insulin signaling transduction that leads to insulin resistance [15]. Consistent with this, JNK1-/- mice are protected from diet-induced insulin resistance, supporting the negative regulatory role of JNK1 on glucose metabolism [14]. However, this interpretation is not without arguments. Hirosumi et al. [14] observed that JNK1-/- mice are also resistant to diet-induced obesity. This is an important observation since the protective effects of JNK1 deficiency against diet-induced insulin resistance can be at least partially explained by the fact that JNK1-/- mice became less obese after HFD. In fact, many transgenic mouse models that are resistant to diet-induced obesity remain more insulin sensitive following HFD due to their lean phenotypes [16,17]. Therapeutic intervention that reduces obesity in HFD-fed mice also results in improved insulin sensitivity [18]. Using various genetic approaches, Roger Davis and his colleagues have extensively investigated the role of JNK1 in the context of insulin resistance in different metabolic organs. Their findings are anything but expected, and provide major advancement in our understanding of the role of JNK1 in inflammation and insulin resistance.
- Adipose tissue is characterized by macrophage infiltration and increased cytokine secretion in obesity [19]. Adipose function of JNK1 was examined in mice with adipocyte-selective deletion of JNK1, which were generated using mice with conditional (floxed) JNK1 and adipose-specific expression of Cre recombinase (Fabp4-Cre) [20]. In these mice, JNK1 was selectively deleted in white and brown adipose tissue, while JNK1 expression was preserved in other organs [20]. Following HFD, adipose JNK1 deficient mice became obese and gained similar fat mass as compared to Cre-expressing wild-type mice. Despite becoming obese, adipose JNK1 deficient mice were more insulin sensitive, based on insulin tolerance test, after HFD [20]. Surprisingly, improved insulin sensitivity was largely due to increased insulin action in liver as shown during the hyperinsulinemic-euglycemic clamp in HFD-fed JNK1 deficient mice [20]. Insulin-stimulated Akt phosphorylation, which is normally reduced in HFD-fed wild-type liver, was increased in JNK1 deficient mice. The mechanism by which adipose deletion of JNK1 protects liver against diet-induced insulin resistance involves adipocyte secretion of IL-6. High-fat feeding markedly elevates IL-6 mRNA and its circulating levels in obese, wild-type mice. However, JNK1 deletion in adipose tissue completely prevented diet-induced adipose secretion of IL-6, and maintained normal insulin action in liver. Thus, these findings indicate that adipose-derived IL-6 is an important mediator of diet-induced insulin resistance in liver, and that JNK1 is an important regulator of this cross-talk between adipose tissue and liver in obesity [20].
JNK1 REGULATES ADIPOSE INFLAMMATION IN OBESITY
- The liver is a major organ of glucose homeostasis. In fasting state, liver produces glucose (hepatic glucose production) by breaking down stored glycogen and synthesizing new glucose from non-carbohydrate source to maintain euglycemia [21]. Following meals, hepatic glucose production is suppressed by insulin that involves insulin signaling pathway [22]. Insulin resistance and excess production of glucose by liver are the primary cause of fasting hyperglycemia in type 2 diabetes [23]. Recent studies using adenoviral delivery of dominant-negative JNK or JNK-shRNA have shown that JNK1 is an important regulator of hepatic glucose metabolism [24,25]. In a recent study of Sabio et al. [26], JNK1 was selectively deleted in hepatocytes using albumin-cre as a promoter, and their findings are again remarkable. A high-fat feeding increased JNK1 activation in wild-type liver, but did not alter JNK1 activity in liver-specific JNK1 deficient mice. In contrast to previously shown negative regulatory role of JNK1 on hepatic insulin signaling using adenovirus approach, cre-lox mediated JNK1 deletion in hepatocytes failed to rescue diet-induced insulin resistance in liver [26]. Instead, JNK1 deficient mice developed insulin resistance and hepatic steatosis on chow-fed state [26]. The hyperinsulinemic-euglycemic clamp showed that this was due to reduced hepatic insulin action and blunted Akt-mediated insulin signaling in liver-specific JNK1 deficient mice. JNK1 deficiency in liver also elevated hepatic expression of genes associated with lipid metabolism (e.g., PGC-1β, PPARγ, FAS, acetyl CoA carboxylase [ACC]) that resulted in hepatic steatosis in these mice [26]. Since non-alcoholic fatty liver disease is a major cause of liver dysfunction and is closely associated with metabolic syndrome [27], this newly identified role of JNK1 in regulating lipid metabolism is critical in understanding a major liver disease. Furthermore, insulin clearance by the liver is an important determinant of circulating insulin levels, and abnormal hepatic clearance of insulin has been shown to modulate peripheral glucose metabolism and obesity [28,29]. In that regard, hepatic JNK1 deficiency resulted in elevated insulin clearance that was compensated by increased insulin secretion [26]. Taken together, these findings indicate an intricate role of JNK1 in the regulation hepatic glucose and lipid metabolism.
PROTECTIVE ROLE OF JNK1 IN HEPATIC STEATOSIS AND INSULIN RESISTANCE
- Skeletal muscle is a major organ of glucose disposal, and insulin resistance in skeletal muscle is a hallmark feature of type 2 diabetes [1]. Skeletal muscle deletion of insulin receptor or glucose transporter (GLUT4) results in insulin resistance [30,31]. In addition to adipose tissue, high-fat feeding was recently shown to increase macrophage infiltration and cause inflammation in skeletal muscle [32]. Obesity-induced macrophage infiltration in muscle was associated with increased local expression of IL-6 and TNF-α [32]. In that regard, both IL-6 and TNF-α are shown to alter insulin signaling and glucose metabolism in skeletal muscle [33,34]. IL-6 was shown to activate STAT3-SOCS3 expression and promote SOCS3-mediated downregulation of insulin signaling in isolated hepatocytes and adipocytes [35,36]. TNF-α was shown to inhibit AMP-activated protein kinase (AMPK) and alter glucose metabolism in skeletal muscle [34]. The role of JNK1 in muscle metabolism was determined in muscle-specific JNK1 deficient mice using muscle creatine kinase (MCK-Cre) promoter [37]. A high-fat feeding caused JNK1 activation in skeletal muscle that was associated with insulin resistance in wild-type mice [37]. In contrast, diet-induced JNK1 activation was not detected in skeletal muscle of JNK1 deficient mice. Despite becoming obese, muscle-JNK1 deficient mice were protected from insulin resistance and showed increased Akt activation and glucose metabolism in skeletal muscle following HFD [37]. Interestingly, muscle JNK1 deletion resulted in increases in circulating levels of triglyceride and hepatic steatosis following HFD [37]. Muscle JNK1 deficiency also enhanced diet-induced inflammation in adipose tissue, which was responsible for blunted Akt activation in adipose tissue and liver after HFD. Therefore, these findings implicate that JNK1 mediates diet-induced insulin resistance in skeletal muscle, and further modulates lipid metabolism that secondarily affects adipose tissue inflammation and hepatic steatosis. This unexpected combination of events in response to muscle JNK1 deficiency again reflects a complex regulation of glucose and lipid metabolism by JNK1.
JNK1 MEDIATES DIET-INDUCED INSULIN RESISTANCE IN SKELETAL MUSCLE
- Of those recently identified cell-autonomous effects of JNK1, none is as intriguing and important as the role of JNK1 in the murine nervous system. Mice with whole body deletion of JNK1 are resistant to diet-induced obesity, suggesting a potential role of JNK1 in energy balance [14]. Since JNK1 deletion in adipose tissue, liver, and skeletal muscle did not affect diet-induced weight gain, a different organ must therefore account for JNK1 regulation of diet-induced obesity. With established functions of hypothalamus and pituitary gland in regulating energy balance [38], brain is an obvious target of JNK1 effects on weight gain following HFD. This precise and logical question was addressed in mice with brain-specific deletion of JNK1 using Nestin-cre promoter [39]. Remarkably, JNK1 deficiency selectively in nervous system exactly recapitulated resistance to diet-induced obesity phenotypes observed in global JNK1-deficient mice [14]. Following HFD, mice with nervous system JNK1 deficiency remained lean as compared to wild-type littermates. Their positive energy balance was due to reduced food intake, and increased whole body energy expenditure in JNK1 deficient mice [39]. As a result of their leanness, nervous system JNK1 deficient mice were more insulin sensitive than wild-type mice after HFD [39]. Interestingly, gene expression analysis showed increased expression of mRNA derived from thyroid hormone target genes in JNK1 deficient mice. Indeed, enhanced energy expenditure in nervous system JNK1 deficient mice was due to increased thyroid hormone levels and their action [39]. Thus, these results indicate that the hypothalamic-pituitary-thyroid axis is an important target of metabolic regulation by nervous system JNK1. Taken together, all of these studies clearly demonstrate that there are cell-autonomous effects of JNK1 in adipose tissue, liver, skeletal muscle, and brain (Fig. 1). These differential and multifaceted effects of JNK1 must be carefully considered in the design of novel JNK1-related therapeutic interventions in the treatment of insulin resistance.
NERVOUS SYSTEM JNK1 REGULATES ENERGY BALANCE VIA HYPOTHALAMIC-PITUITARY-THYROID AXIS
- A molecular scaffold, kinase suppressor of Ras 2 (KSR2) is a critical regulator of energy balance and glucose homeostasis [40]. In both cell culture and animal models, KSR2 deficiency results in impaired energy expenditure, reduced glucose and lipid metabolism, obesity, and insulin resistance [40]. The underlying mechanism involves KSR2 regulation of AMPK, which is an important sensor of cell's nutritional status and master regulator of systemic energy balance [41]. In response to cellular stress associated with rising AMP or falling ATP, AMPK is activated and increases glucose and lipid metabolism [42]. KSR2 is shown to directly interact with AMPKα1 subunit and promotes phosphorylation of AMPK on Thr172, an essential step in AMPK signaling. The end result is inhibitory phosphorylation of ACC, a rate-limiting step in the conversion of acetyl-CoA to malonyl CoA, and low malonyl CoA level relieves its inhibition of carnitine:palmitoyl-CoA transferase-1 (CPT1), a rate-controlling step in mitochondrial fatty acid oxidation [43, 44]. AMPK also increases glucose transport and glycogen metabolism to further restore cellular energy needs [45]. Consistent with this, KSR2 deficient mice showed impaired energy expenditure and became obese. KSR2 deficient mice also developed insulin resistance in multiple organs including skeletal muscle, liver, and adipose tissue, which may be due to cell-autonomous effects of KSR2 on glucose and lipid metabolism and obesity [40].
- As a master regulator of cellular metabolism, AMPK acts on multiple organs including skeletal muscle, adipose tissue, liver, heart and brain, and exerts diverse effects to maintain energy homeostasis [41]. Alterations in metabolism and energy expenditure are hallmark features of obesity and type 2 diabetes, and therapeutic activator of AMPK is actively investigated to treat metabolic disease and its complications [46,47]. Thus, the current findings on KSR2 regulation of AMPK unveil an exciting new pathway to regulate energy balance and potential drug targets (Fig. 2). More importantly, this study unveils novel insights into the role of molecular scaffold in energy homeostasis. In that regard, JNK interacting protein 1 is another scaffold protein that regulates stress kinase signaling such as JNK1 and is involved in glucose metabolism [13,48]. This is yet another evidence that other molecular scaffold proteins in the regulation of insulin resistance remain to be discovered.
MOLECULAR SCAFFOLD KSR2 REGULATES ENERGY BALANCE AND GLUCOSE METABOLISM
- The endoplasmic reticulum (ER) is a specialized perinuclear organelle for the synthesis of secretory and membrane-targeted proteins. ER stress results from an imbalance between protein load and folding capacity that leads to unfolded protein response (UPR) [49]. The UPR activates 3 major ER signaling pathways: 1) PKR-like endoplasmic reticulum kinase, 2) inositol requiring-1 (IRE-1), and 3) activating transcription factor 6 [50]. In obese state, intracellular lipid accumulation activates IRE-1 and the stress kinase signaling such as JNK1 [14]. Treatment with chemical chaperones such as 4-phenyl butyric acid or tauroursodeoxycholic acid has been shown to attenuate ER stress and improve insulin sensitivity in diet-induced obese mice [51]. These observations implicate an important role of ER homeostasis in obesity and insulin resistance.
- The 78-kDa glucose regulated protein, GRP78, also known as BiP (immunoglobulin heavy-chain binding protein) or HSPA5, is a key rheostat in regulating ER homeostasis [52]. GRP78 regulates ER function via protein folding and assembly, targeting misfolded protein for degradation, ER Ca2+ binding, and controlling the activation of transmembrane ER stress sensors [52]. To determine the role of ER stress in obesity, mice with heterozygous deletion of Grp78 were recently developed and shown to be resistant to diet-induced obesity [53]. Grp78 deficient mice showed enhanced whole body energy expenditure, and were more insulin sensitive as compared to wild-type littermates following HFD [53]. This unexpected finding in which Grp78 heterozygosity improves energy balance and insulin sensitivity is due to remarkable activation of adaptive UPR, which resulted in improved ER homeostasis in adipose tissue [53]. Thus, these findings support the important role of ER homeostasis and ER stress in energy balance and the regulation of glucose metabolism.
ENDOPLASMIC RETICULUM CHAPERONE GRP78 REGULATES ENERGY BALANCE AND INSULIN RESISTANCE
- If inflammation develops in white adipose tissue, liver, and skeletal muscle, what other organs are affected by inflammation in obesity? Herrero et al. [54] recently showed that brown adipose tissue develops macrophage infiltration and inflammation in lipodystrophic mice. A recent study from our group found that diet-induced obesity causes macrophage infiltration in heart, and elevated IL-6 levels suppress AMPK activity and myocardial glucose metabolism [55]. It will be important to determine what other organs develop inflammation, and how they may contribute to insulin resistance in obesity.
- If inflammatory cytokines such as IL-6 and TNF-α mediate insulin resistance, are there cytokines that oppose the action of IL-6 and TNF-α and positively regulate insulin sensitivity? In that regard, IL-10 is a potent inhibitor of the pro-inflammatory cytokines and chemokines [56]. Plasma IL-10 levels are positively related to insulin sensitivity in healthy subjects, and IL-10 levels are reduced in obese, diabetic subjects [57,58]. A recent report from Lumeng et al. [59] demonstrated that adipose tissue macrophages from lean animals express polarization toward an alternatively activated state, and this was associated with increased expression of IL-10. This study further showed that IL-10 increases glucose uptake and protects against TNF-α mediated insulin resistance in isolated adipocytes [59]. Moreover, our recent study found that elevation of IL-10 levels by chronic IL-10 treatment or transgenic overexpression of IL-10 improves insulin sensitivity in skeletal muscle following HFD [32]. It will be important to further understand the metabolic role of IL-10 and discover other cytokines that may regulate insulin resistance.
FUTURE QUESTIONS
- It is not presumptuous to state that diabetes is an inflammatory disease. At the cellular level, it involves a complex network of stress kinase signaling, molecular scaffolds, and ER chaperones. At the systemic level, cytokines play a major role in cross-talk between organs. Diabetes is neither an energy deficient nor energy surplus state. Instead, it is a state of energy imbalance. In the past decade, we have acquired a greater understanding of molecular events and signaling pathways to link inflammation and insulin resistance. They are new exciting ideas for an age-old question.
CONCLUSION
- 1. Kahn CR. Banting lecture: insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 1994;43:1066-1084. PubMed
- 2. Reaven GM. Banting lecture 1988: role of insulin resistance in human disease. Diabetes 1988;37:1595-1607. ArticlePubMed
- 3. Boden G. Obesity, free fatty acids, and insulin resistance. Curr Opin Endocrinol Diabetes 2001;8:235-239.Article
- 4. McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science 1992;258:766-770. ArticlePubMed
- 5. Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000;106:171-176. ArticlePubMedPMC
- 6. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87-91. ArticlePubMed
- 7. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994;372:425-432. ArticlePubMedPDF
- 8. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 1995;270:26746-26749. ArticlePubMed
- 9. Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acutephase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997;40:1286-1292. ArticlePubMedPDF
- 10. Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 2001;280:E745-E751. ArticlePubMed
- 11. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 2005;115:1111-1119. ArticlePubMedPMC
- 12. Williamson RT. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. Br Med J 1901;1:760-762.ArticlePubMedPMC
- 13. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol 2007;19:142-149. ArticlePubMed
- 14. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature 2002;420:333-336. ArticlePubMedPDF
- 15. Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 2000;275:9047-9054. ArticlePubMed
- 16. Dubois MJ, Bergeron S, Kim HJ, Dombrowski L, Perreault M, Fournes B, Faure R, Olivier M, Beauchemin N, Shulman GI, Siminovitch KA, Kim JK, Marette A. The SHP-1 protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat Med 2006;12:549-556. ArticlePubMedPDF
- 17. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G. Endocrine regulation of energy metabolism by the skeleton. Cell 2007;130:456-469. ArticlePubMedPMC
- 18. Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, Chen JL, Jung DY, Zhang Z, Ko HJ, Kim JK, Veniant MM. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009;58:250-259. ArticlePubMedPMCPDF
- 19. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821-1830. ArticlePubMedPMC
- 20. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008;322:1539-1543. ArticlePubMedPMC
- 21. DeFronzo RA, Ferrannini E. Regulation of hepatic glucose metabolism in humans. Diabetes Metab Rev 1987;3:415-459. ArticlePubMed
- 22. DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM: a balanced overview. Diabetes Care 1992;15:318-368. ArticlePubMedPDF
- 23. Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 2000;6:87-97. ArticlePubMed
- 24. Nakatani Y, Kaneto H, Kawamori D, Hatazaki M, Miyatsuka T, Matsuoka TA, Kajimoto Y, Matsuhisa M, Yamasaki Y, Hori M. Modulation of the JNK pathway in liver affects insulin resistance status. J Biol Chem 2004;279:45803-45809. ArticlePubMed
- 25. Yang R, Wilcox DM, Haasch DL, Jung PM, Nguyen PT, Voorbach MJ, Doktor S, Brodjian S, Bush EN, Lin E, Jacobson PB, Collins CA, Landschulz KT, Trevillyan JM, Rondinone CM, Surowy TK. Liver-specific knockdown of JNK1 up-regulates proliferator-activated receptor gamma coactivator 1 beta and increases plasma triglyceride despite reduced glucose and insulin levels in diet-induced obese mice. J Biol Chem 2007;282:22765-22774. PubMed
- 26. Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK, Davis RJ. Prevention of steatosis by hepatic JNK1. Cell Metab 2009;10:491-498. ArticlePubMedPMC
- 27. Angulo P, Lindor KD. Treatment of non-alcoholic steatohepatitis. Best Pract Res Clin Gastroenterol 2002;16:797-810. ArticlePubMed
- 28. Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev 1998;19:608-624. ArticlePubMed
- 29. Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab 2002;13:240-245. ArticlePubMed
- 30. Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, Kahn BB, Kahn CR, Shulman GI. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest 2000;105:1791-1797. ArticlePubMedPMC
- 31. Kim JK, Zisman A, Fillmore JJ, Peroni OD, Kotani K, Perret P, Zong H, Dong J, Kahn CR, Kahn BB, Shulman GI. Glucose toxicity and the development of diabetes in mice with muscle-specific inactivation of GLUT4. J Clin Invest 2001;108:153-160. ArticlePubMedPMC
- 32. Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, Friedline RH, Kurt-Jones E, Finberg R, Fischer MA, Granger EL, Norbury CC, Hauschka SD, Philbrick WM, Lee CG, Elias JA, Kim JK. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 2009;58:2525-2535. ArticlePubMedPMCPDF
- 33. Kim HJ, Higashimori T, Park SY, Choi H, Dong J, Kim YJ, Noh HL, Cho YR, Cline G, Kim YB, Kim JK. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 2004;53:1060-1067. ArticlePubMedPDF
- 34. Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Gorgun CZ, Carling D, Hotamisligil GS, Febbraio MA, Kay TW, Kemp BE. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab 2006;4:465-474. PubMed
- 35. Shi H, Tzameli I, Bjorbaek C, Flier JS. Suppressor of cytokine signaling 3 is a physiological regulator of adipocyte insulin signaling. J Biol Chem 2004;279:34733-34740. ArticlePubMed
- 36. Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 2002;277:42394-42398. ArticlePubMed
- 37. Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK, Davis RJ. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol 2010;30:106-115. ArticlePubMedPDF
- 38. Lenard NR, Berthoud HR. Central and peripheral regulation of food intake and physical activity: pathways and genes. Obesity (Silver Spring) 2008;16(Suppl 3):S11-S22. ArticlePMCPDF
- 39. Sabio G, Cavanagh-Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, Morel C, Mora A, Reilly J, Kim JK, Davis RJ. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev 2010;24:256-264. ArticlePubMedPMC
- 40. Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O, Fernandez MR, Fisher K, Kortum RL, Hong EG, Jun JY, Ko HJ, Schreiner A, Volle DJ, Treece T, Swift AL, Winer M, Chen D, Wu M, Leon LR, Shaw AS, McNeish J, Kim JK, Morrison DK, Tschop MH, Lewis RE. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab 2009;10:366-378. ArticlePubMedPMC
- 41. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005;1:15-25. ArticlePubMed
- 42. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2007;8:774-785. ArticlePubMedPDF
- 43. Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 2004;3:340-351. ArticlePubMedPDF
- 44. Ruderman NB, Saha AK, Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology 2003;144:5166-5171. ArticlePubMed
- 45. Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Mol Cell Biochem 2003;253:65-70. PubMed
- 46. Musi N, Goodyear LJ. Targeting the AMP-activated protein kinase for the treatment of type 2 diabetes. Curr Drug Targets Immune Endocr Metabol Disord 2002;2:119-127. ArticlePubMed
- 47. Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest 2004;114:495-503. ArticlePubMedPMC
- 48. Jaeschke A, Czech MP, Davis RJ. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev 2004;18:1976-1980. ArticlePubMedPMC
- 49. Kaufman RJ, Scheuner D, Schroder M, Shen X, Lee K, Liu CY, Arnold SM. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol 2002;3:411-421. ArticlePubMedPDF
- 50. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003;23:7448-7459. ArticlePubMedPMCPDF
- 51. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006;313:1137-1140. ArticlePubMedPMC
- 52. Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci 2001;26:504-510. ArticlePubMed
- 53. Ye R, Jung DY, Jun JY, Li J, Luo S, Ko HJ, Kim JK, Lee AS. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 2010;59:6-16. ArticlePubMedPDF
- 54. Herrero L, Shapiro H, Nayer A, Lee J, Shoelson SE. Inflammation and adipose tissue macrophages in lipodystrophic mice. Proc Natl Acad Sci U S A 2010;107:240-245. ArticlePubMed
- 55. Ko HJ, Zhang Z, Jung DY, Jun JY, Ma Z, Jones KE, Chan SY, Kim JK. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes 2009;58:2536-2546. ArticlePubMedPMCPDF
- 56. Akdis CA, Blaser K. Mechanisms of interleukin-10-mediated immune suppression. Immunology 2001;103:131-136. ArticlePubMedPMC
- 57. Straczkowski M, Kowalska I, Nikolajuk A, Krukowska A, Gorska M. Plasma interleukin-10 concentration is positively related to insulin sensitivity in young healthy individuals. Diabetes Care 2005;28:2036-2037. ArticlePubMedPDF
- 58. Esposito K, Pontillo A, Giugliano F, Giugliano G, Marfella R, Nicoletti G, Giugliano D. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J Clin Endocrinol Metab 2003;88:1055-1058. ArticlePubMedPDF
- 59. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175-184. ArticlePubMedPMC
REFERENCES
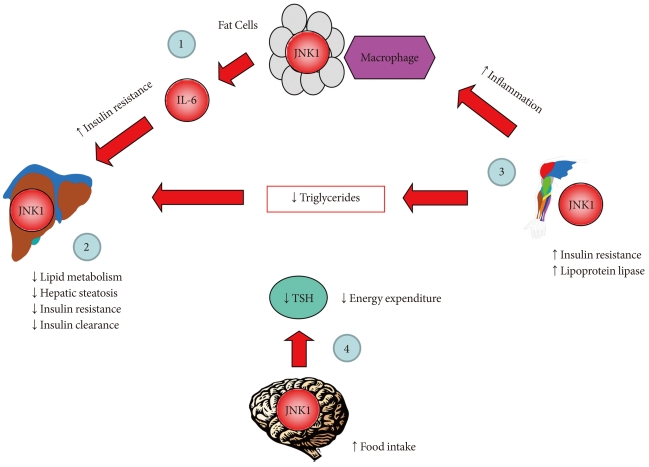
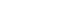
Fig. 1cJun NH2-terminal kinase 1 (JNK1) regulates energy balance and glucose and lipid homeostasis via cell-autonomous manner. Based on the findings from mice with JNK1 deficiency selectively in adipose tissue, liver, skeletal muscle, or nervous system: 1) adipose tissue JNK1 promotes interleukin-6 (IL-6) secretion which causes hepatic insulin resistance in obesity, 2) liver JNK1 reduces lipid metabolism and insulin clearance thereby preventing hepatic steatosis and insulin resistance, 3) skeletal muscle JNK1 mediates insulin resistance, adipose tissue inflammation, and suppresses muscle lipoprotein lipase thereby altering circulating triglyceride levels, and 4) nervous system JNK1 mediates the negative feedback regulation of hypothalamic-pituitary-thyroid axis and promotes negative energy balance by increasing food intake and reducing energy expenditure.
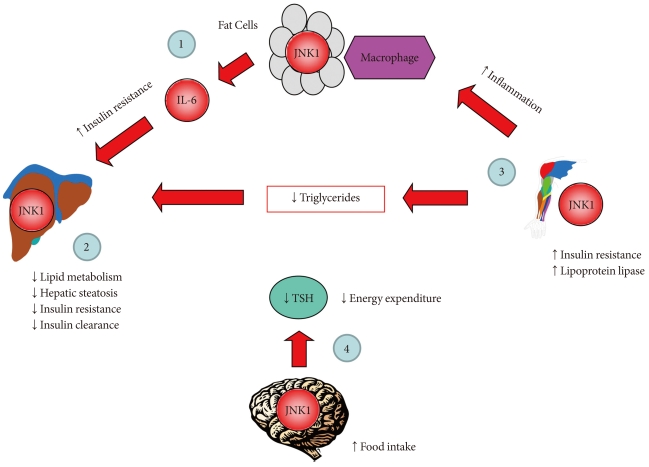
Fig. 2Kinase suppressor of Ras 2 (KSR2) regulates obesity and insulin resistance by activating AMP-activated protein kinase (AMPK). KSR2 regulates lipid metabolism by activating AMPK, which phosphorylates and inactivates acetyl CoA carboxylase (ACC), and this leads to reduced malonyl CoA level that relieves its inhibition of carnitine:palmitoyl-CoA transferase-1 (CPT1), a rate-controlling step in mitochondrial fatty acid oxidation. As a result, KSR2 increases lipid oxidation and reduces lipid storage and obesity. KSR2-mediated AMPK activation also increases glucose metabolism and insulin action.
Figure & Data
References
Citations
Citations to this article as recorded by 
- HMC Ameliorates Hyperglycemia via Acting PI3K/AKT Pathway and Improving FOXO1 Pathway in ob/ob Mice
Jeong Yoo, Jae Eun Park, Ji Sook Han
Nutrients.2023; 15(9): 2023. CrossRef - Licochalcone A: A Potential Multitarget Drug for Alzheimer’s Disease Treatment
Jordi Olloquequi, Miren Ettcheto, Amanda Cano, Ana Fortuna, Joana Bicker, Elena Sánchez-Lopez, Cristian Paz, Jesús Ureña, Ester Verdaguer, Carme Auladell, Antoni Camins
International Journal of Molecular Sciences.2023; 24(18): 14177. CrossRef - HM-Chromanone, a Major Homoisoflavonoid in Portulaca oleracea L., Improves Palmitate-Induced Insulin Resistance by Regulating Phosphorylation of IRS-1 Residues in L6 Skeletal Muscle Cells
Jae-Eun Park, Ji-Sook Han
Nutrients.2022; 14(18): 3815. CrossRef - Assessment of PON1 activity and circulating TF levels in relation to BMI, testosterone, HOMA-IR, HDL-C, LDL-C, CHO, SOD activity and TAC in women with PCOS: An observational study
Humira Jeelani, Mohd Ashraf Ganie, Akbar Masood, Shajrul Amin, Iram Ashaq Kawa, Qudsia Fatima, Saika Manzoor, Tabasum Parvez, Niyaz Ahmad Naikoo, Fouzia Rashid
Diabetes & Metabolic Syndrome: Clinical Research & Reviews.2019; 13(5): 2907. CrossRef - Metformin and sulodexide restore cardiac microvascular perfusion capacity in diet-induced obese rats
Judith van Haare, M. Eline Kooi, Jurgen W. G. E. van Teeffelen, Hans Vink, Jos Slenter, Hanneke Cobelens, Gustav J. Strijkers, Dennis Koehn, Mark J. Post, Marc van Bilsen
Cardiovascular Diabetology.2017;[Epub] CrossRef - CEACAM1 loss links inflammation to insulin resistance in obesity and non-alcoholic steatohepatitis (NASH)
Sonia M. Najjar, Lucia Russo
Seminars in Immunopathology.2014; 36(1): 55. CrossRef - Immunoglobulin E and mast cell proteases are potential risk factors of impaired fasting glucose and impaired glucose tolerance in humans
Zhen Wang, Hong Zhang, Xu-Hui Shen, Kui-Li Jin, Guo-fen Ye, Wei Qiu, Li Qian, Bo Li, Yong-Hong Zhang, Guo-Ping Shi
Annals of Medicine.2013; 45(3): 220. CrossRef - Reduction of Insulin Resistance and Plasma Glucose Level by Salsalate Treatment in Persons With Prediabetes
Elham Faghihimani, Ashraf Aminorroaya, Hassan Rezvanian, Peyman Adibi, Faramarz Ismail-Beigi, Masoud Amini
Endocrine Practice.2012; 18(6): 826. CrossRef - Immunoglobulin E and Mast Cell Proteases Are Potential Risk Factors of Human Pre-Diabetes and Diabetes Mellitus
Zhen Wang, Hong Zhang, Xu-Hui Shen, Kui-Li Jin, Guo-fen Ye, Li Qian, Bo Li, Yong-Hong Zhang, Guo-Ping Shi, Yiqing Song
PLoS ONE.2011; 6(12): e28962. CrossRef
 PubReader
PubReader Cite
Cite