- Skip Navigation
- Skip to contents

- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 48(1); 2024 > Article
-
Original ArticleBasic Research Glucagon-Like Peptide Receptor Agonist Inhibits Angiotensin II-Induced Proliferation and Migration in Vascular Smooth Muscle Cells and Ameliorates Phosphate-Induced Vascular Smooth Muscle Cells Calcification
-
Jinmi Lee1*
 , Seok-Woo Hong1*, Min-Jeong Kim1, Sun Joon Moon2, Hyemi Kwon2, Se Eun Park2, Eun-Jung Rhee2, Won-Young Lee2
, Seok-Woo Hong1*, Min-Jeong Kim1, Sun Joon Moon2, Hyemi Kwon2, Se Eun Park2, Eun-Jung Rhee2, Won-Young Lee2 -
Diabetes & Metabolism Journal 2024;48(1):83-96.
DOI: https://doi.org/10.4093/dmj.2022.0363
Published online: January 3, 2024
1Institute of Medical Research, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea
2Division of Endocrinology and Metabolism, Department of Internal Medicine, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea
-
Corresponding authors: Won-Young Lee Division of Endocrinology and Metabolism, Department of Internal Medicine, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, 29 Saemunan-ro, Jongno-gu, Seoul 03181, Korea E-mail: wonyoung2.lee@samsung.com
-
Eun-Jung Rhee Division of Endocrinology and Metabolism, Department of Internal Medicine, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, 29 Saemunan-ro, Jongno-gu, Seoul 03181, Korea E-mail: ejrhee.lee@samsung.com
- *Jinmi Lee and Seok-Woo Hong contributed equally to this study as first authors.
Copyright © 2024 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Glucagon-like peptide-1 receptor agonist (GLP-1RA), which is a therapeutic agent for the treatment of type 2 diabetes mellitus, has a beneficial effect on the cardiovascular system.
-
Methods
- To examine the protective effects of GLP-1RAs on proliferation and migration of vascular smooth muscle cells (VSMCs), A-10 cells exposed to angiotensin II (Ang II) were treated with either exendin-4, liraglutide, or dulaglutide. To examine the effects of GLP-1RAs on vascular calcification, cells exposed to high concentration of inorganic phosphate (Pi) were treated with exendin-4, liraglutide, or dulaglutide.
-
Results
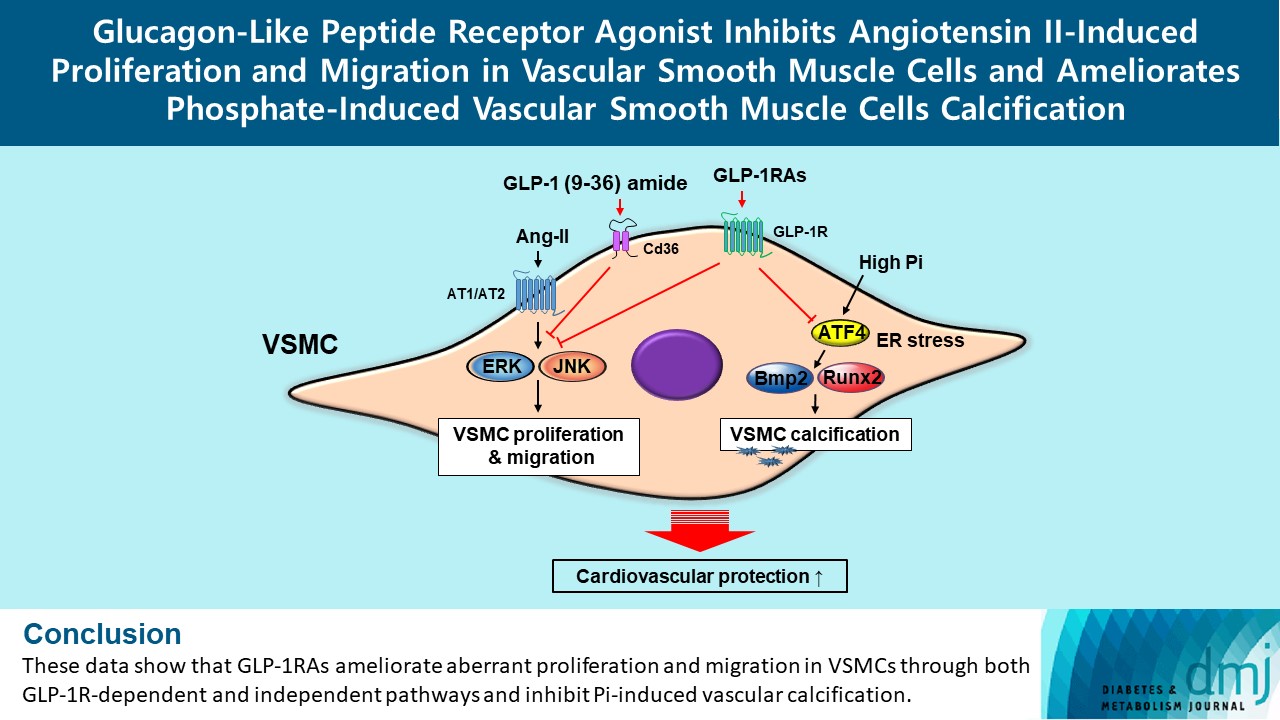
- Ang II increased proliferation and migration of VSMCs, gene expression levels of Ang II receptors AT1 and AT2, proliferation marker of proliferation Ki-67 (Mki-67), proliferating cell nuclear antigen (Pcna), and cyclin D1 (Ccnd1), and the protein expression levels of phospho-extracellular signal-regulated kinase (p-Erk), phospho-c-JUN N-terminal kinase (p-JNK), and phospho-phosphatidylinositol 3-kinase (p-Pi3k). Exendin-4, liraglutide, and dulaglutide significantly decreased the proliferation and migration of VSMCs, the gene expression levels of Pcna, and the protein expression levels of p-Erk and p-JNK in the Ang II-treated VSMCs. Erk inhibitor PD98059 and JNK inhibitor SP600125 decreased the protein expression levels of Pcna and Ccnd1 and proliferation of VSMCs. Inhibition of GLP-1R by siRNA reversed the reduction of the protein expression levels of p-Erk and p-JNK by exendin-4, liraglutide, and dulaglutide in the Ang II-treated VSMCs. Moreover, GLP-1 (9-36) amide also decreased the proliferation and migration of the Ang II-treated VSMCs. In addition, these GLP-1RAs decreased calcium deposition by inhibiting activating transcription factor 4 (Atf4) in Pi-treated VSMCs.
-
Conclusion
- These data show that GLP-1RAs ameliorate aberrant proliferation and migration in VSMCs through both GLP-1R-dependent and independent pathways and inhibit Pi-induced vascular calcification.
- Keywords: Angiotensin II; Cell migration inhibition; Cell proliferation; Glucagon-like peptide 1; Muscle, smooth, vascular
- Cardiovascular disease (CVD) includes all disorders of the heart and blood vessels, such as coronary heart disease, ischemic heart disease, hypertension, stroke, and atherosclerosis, and is a major cause of mortality and morbidity among patients with diabetes [1,2]. The risk for CVD morbidity and mortality is 2 to 4-fold higher in patients with type 2 diabetes mellitus (T2DM) than in those without diabetes, and the prevalence of T2DM is steadily increasing worldwide [3].
- Atherosclerosis is a chronic disease associated with the interaction between lipid metabolism and inflammation [4,5]. The atherosclerotic process is initiated by the accumulation of low-density lipoprotein (LDL) cholesterol, which causes vascular smooth muscle cell (VSMC) injury. In atherosclerosis, lipid accumulation induces transdifferentiation of VSMCs into alternative phenotypes such as macrophage-like, foam cell-like, and osteochondrogenic-like, which contribute to plaque progression [6,7]. In addition, VSMCs are involved in all developmental and progression stages of atherosclerosis, through migration, proliferation, and apoptosis. Aberrant proliferation and migration of VSMCs following phenotypic switching are hallmarks of atherosclerosis [4,8].
- Angiotensin II (Ang II) is the effector peptide of the reninangiotensin system and is implicated in endothelial dysfunction, atherosclerosis, hypertension, and related vascular diseases [9]. The effects of Ang II are mediated through two receptor subtypes: the angiotensin II receptor type 1 (AT1), which is involved in inflammation, proliferation hypertrophy, and vasoconstriction; and the angiotensin II receptor type 2 (AT2), which is involved in apoptosis, hypotension, and vasodilation [10,11]. The AT2 receptor inhibits the AT1 receptor [12,13], and the ratio of AT1 to AT2 is closely related to the cardiovascular complications associated with diabetes [14]. In addition, AT1 antagonists inhibit VSMC proliferation and migration via the adenosine monophosphate-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) signaling pathway [15].
- Vascular calcification is the deposition of minerals in the artery walls, and it is closely related to CVDs [16,17]. Previous studies reported that intimal calcification is associated with atherosclerosis, while medial calcification is associated with diabetes, chronic kidney disease, and aging [18,19]. VSMCs are the most abundant component of the middle layer of the artery wall, and the transdifferentiation of VSMCs by a high concentration of inorganic phosphate (Pi) is one of the major pathways leading to medial calcification [20].
- Glucagon-like peptide-1 receptor agonists (GLP-1RAs) are glucose-lowering medications used in the treatment of T2DM. GLP-1 receptor (GLP-1R) expression is detected in various cardiovascular tissues [21]. A few GLP-1RAs, such as liraglutide and semaglutide, have been shown to effectively improve cardiovascular outcomes in patients with T2DM in the Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial and the Trial to Evaluate Cardiovascular and Other Long-term Outcomes with Semaglutide in Subjects with Type 2 Diabetes (SUSTAIN-6) trial, respectively [22,23]. Previous studies reported that dipeptidyl peptidase-4 (DPP-4) rapidly metabolizes GLP-1 into GLP-1 (9-36) amide, resulting in the loss of its insulinotropic activity [24,25]. However, recent studies reported that GLP-1 (9-36) amide has a GLP-1R-independent cardioprotective action [26,27].
- In the present study, we investigated the protective effects and regulatory mechanism of GLP-1RAs on Ang II-induced VSMC proliferation and migration and Pi-induced VSMC calcification.
INTRODUCTION
- Cell culture
- The rat aortic A-10 VSMC line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and was cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose (25 mmol/L, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA) and 1% penicillin/streptomycin (Gibco) in an incubator at 37°C with a humidified atmosphere of 5% CO2 in the air. To examine the protective effects of GLP-1RAs on proliferation and migration of VSMCs induced by Ang II (Sigma-Aldrich, St. Louis, MO, USA), the cells were pretreated with either 100 nM exendin-4 (Sigma-Aldrich), liraglutide, or dulaglutide (MedChemExpress, Monmouth Junction, NJ, USA) 1 hour before Ang II treatment. During treatment with Ang II and GLP-1RAs, the cells were incubated in DMEM supplemented with 0.5% FBS (Hyclone). To examine the effects of GLP-1RAs on vascular calcification, cells exposed to 4 mM Pi (Sigma-Aldrich) were treated with 100 nM exendin-4, liraglutide, or dulaglutide for 7 days. The media was changed every 2 to 3 days. Lipopolysaccaharide (Merck Millipore, Burlington, MA, USA) and thapsigargin (Sigma-Aldrich) were treated to activate the mitogen-activated protein kinase (MAPK) pathway and endoplasmic reticulum (ER) stress, respectively.
- Cell proliferation assay
- For analysis of VSMC proliferation, cell viability was determined using an MTT cell proliferation assay kit (Invitrogen, Carlsbad, CA, USA). Cells were seeded in 96 well plates and incubated until 80% confluent. After serum starvation for 6 hours, cells were treated with various concentrations of chemicals for 24 hours. The cell culture medium was removed and replaced with 100 µL fresh culture medium, and 10 µL of the 12 mM MTT stock solution was added. The plate was incubated at 37°C for 4 hours, following which 100 µL of sodium dodecyl sulfate in 0.01 N HCl was added, and the absorbance was read at 450 nm using a microplate reader (TECAN, Grödig, Austria).
- Transwell migration assay
- VSMC migration was determined using the transwell migration assay as previously described [28]. Briefly, the confluent monolayers of VSMCs in 6-well plates were treated with of 1 µM Ang II in the absence or presence of GLP-1RAs at 37°C for 16 hours. The cells were then trypsinized with 0.25% (v/v) trypsin, resuspended in serum-free DMEM, and seeded in the upper chamber of each transwell. In the lower chamber of each transwell, DMEM supplemented with 10% FBS was added as a chemoattractant. After incubation for 24 hours, the upper chamber was fixed with 4% (w/v) formaldehyde for 20 minutes at room temperature, washed with phosphate-buffered saline (PBS), and stained with crystal violet (Sigma-Aldrich) for 15 minutes. After washing with distilled water, the non-migrated cells from the upper surface of the transwell membrane were removed using a cotton swab, and images were captured by microscopy at ×100 magnification (Olympus IX71, Olympus, Tokyo, Japan).
- Scratch wound healing assay
- In addition to the transwell migration assay, VSMC migration was also determined using the scratch wound healing assay. VSMCs were seeded in 6-well plates and when the cells reached 80% to 90% confluence, they were scratched using 200 µL yellow tips in the center of each well. After washing with PBS, the cells were incubated with the chemicals indicated in each experiment. Images were captured at 0 hour and at the time point indicated in the figures (×100 magnification). VSMC migration was quantified by measuring the distance of the scratch wound at both time points using ImageJ and expressed as follows: % gap distance=[(distance at 0 hour time points–distance at the indicated time points)/distance at 0 hour time points]×100.
- Immunofluorescence staining
- VSMCs were cultured on coverslips, fixed with 4% formaldehyde for 5 minutes, permeabilized with pre-chilled 0.5% Triton X-100 for 5 minutes at –20°C, and incubated with a normal goat serum blocking solution for 1 hour at room temperature. The cells were incubated overnight with the primary anti-proliferating cell nuclear antigen (Pcna) antibody at 4°C and were then labeled with a tetramethylrhodamine isothiocyanate-conjugated secondary antibody for 1 hour at room temperature. Next, the cells were mounted using a mounting medium with 4´,6-diamidino-2-phenylindole (DAPI; ab104139, Abcam, Cambridge, UK), and images were obtained using a fluorescence microscope (BX51, Olympus).
- Quantitative real-time polymerase chain reaction
- The total RNA was extracted from VSMCs using the TRIzol reagent (Invitrogen) and cDNA was synthesized via reversetranscription using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. Quantitative real-time polymerase chain reaction (PCR) for target gene expression was performed with LightCycler 480 SYBR Green (Roche, Lewis, UK). The primer pairs used for PCR were purchased from Bioneer (Daejeon, Korea). The primer sequences are presented in Supplementary Table 1. The expression levels of the target genes were normalized to the internal control gene (glyceraldehyde-3-phosphate dehydrogenase [Gapdh]) and calculated using the 2−ΔΔCt method.
- Calcium deposition assay
- To determine the calcium content, cells were incubated with 0.5 N HCl for 24 hours at 4°C. The decalcified solution was measured using the QuantiChrom Calcium Assay Kit (BioAssay Systems, Hayward, CA, USA) and the calcium content was normalized to the total protein concentration. To determine the calcium deposition in the extracellular matrix, the cells were washed with PBS, fixed with 10% formalin for 10 minutes at room temperature, and stained with 0.5% alizarin red S solution for 30 minutes at 37°C. The cells were then washed twice with PBS and photographed using a microscope (Olympus).
- Western blotting
- The cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology, Danvers, MA, USA) containing the Halt Protease and Phosphatase Inhibitor Cocktail (100×) (Thermo Fisher Scientific, Rockford, IL, USA), and the concentration of protein was calculated using Bradford protein analysis (Bio-Rad, Hercules, CA, USA). For Western blotting, protein samples (20 µg) were dissolved in 4× lithium dodecyl sulfate sample buffer and 10× reducing sample agent (Invitrogen), heated at 95°C for 10 minutes, and loaded on a 4% to 12% Bis-Tris NuPAGE gel (Thermo Fisher Scientific). The separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane using the iBlot2 PVDF stack (Invitrogen), and the membranes were blocked with 5% bovine serum albumin in tris-buffered saline with Tween-20 (TBST) for 1 hour at room temperature, following which they were incubated at 4°C overnight with the following specific primary antibodies (Supplementary Table 2). After washing with TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Protein bands were visualized by enhanced chemiluminescence reagent (DAWINBio, Hanam, Korea).
- Transient siRNA transfection
- The A-10 cells were transfected with GLP-1R or activating transcription factor 4 (Atf4) siRNA (Bioneer) using Lipofectamine RNAi MAX transfection reagent (Invitrogen), according to the manufacturer’s instructions. Scrambled siRNA was used as a negative control for transfection. After 24 hours of GLP-1R transfection, the cells were treated with 1 μM Ang II in the presence or absence of various concentration of GLP-1 (9-36) amide (MedChemExpress) or either 100 nM exendin-4, liraglutide, or dulaglutide for 24 hours and harvested for further analysis. After 24 hours of Atf4 siRNA transfection, the cells were treated with 4 mM Pi in the presence or absence of 100 nM exendin-4, liraglutide, or dulaglutide and harvested for further analysis.
- Statistical analysis
- All data were collected in triplicate and are expressed as mean±standard error values. The statistical analysis was performed using SPSS version 12.0 software (SPSS Inc., Chicago, IL, USA). The data were analyzed using one-way analysis of variance, followed by post hoc Bonferroni multiple range tests to calculate significant differences between the mean values obtained from the different experimental groups. A P value of <0.05 was considered statistically significant.
METHODS
- Ang II induces migration and proliferation in VSMCs
- Excessive increase in VSMC proliferation and migration are important hallmarks of atherosclerosis, and Ang II, as well as some types of cytokines and growth factors, stimulate proliferation and migration in VSMCs [9]. Exposure of the A-10 cells to different concentrations of Ang II produced an increase in migration and proliferation of VSMCs, which were observed using the scratch wound healing assay and MTT, respectively (Supplementary Fig. 1A and B). In addition, the expression of Ang II receptors, AT1 and AT2, was increased by Ang II in a dose-dependent manner (Supplementary Fig. 1C). Thus, these results indicate that Ang II induces VSMC proliferation and migration via AT1 and AT2, and 1 µM Ang II should be used as an appropriate concentration to induce VSMC proliferation and migration in further studies.
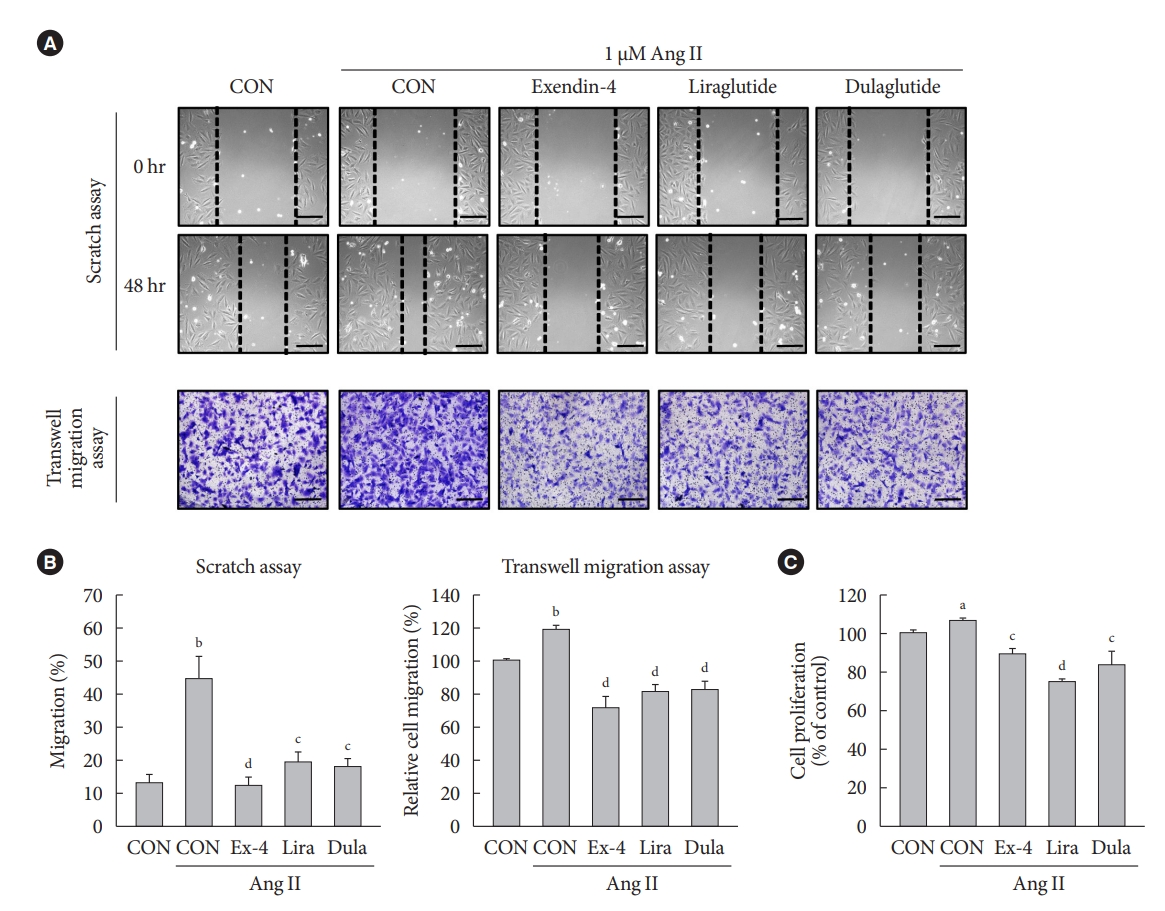
- GLP-1RAs inhibit Ang II-induced migration and proliferation of VSMCs
- To examine the effects of GLP-1RAs on Ang II-induced VSMC migration and proliferation, A-10 cells treated with Ang II were exposed to exendin-4, liraglutide, and dulaglutide. GLP-1RAs significantly decreased Ang II-induced VSMC migration, which was demonstrated by the scratch wound healing and transwell migration assays (Fig. 1A and B). In addition, GLP-1RAs inhibited the proliferation of Ang II-treated cells (Fig. 1C). These results indicate that GLP-1RAs reverse the Ang II-induced increase in migration and proliferation of VSMCs.
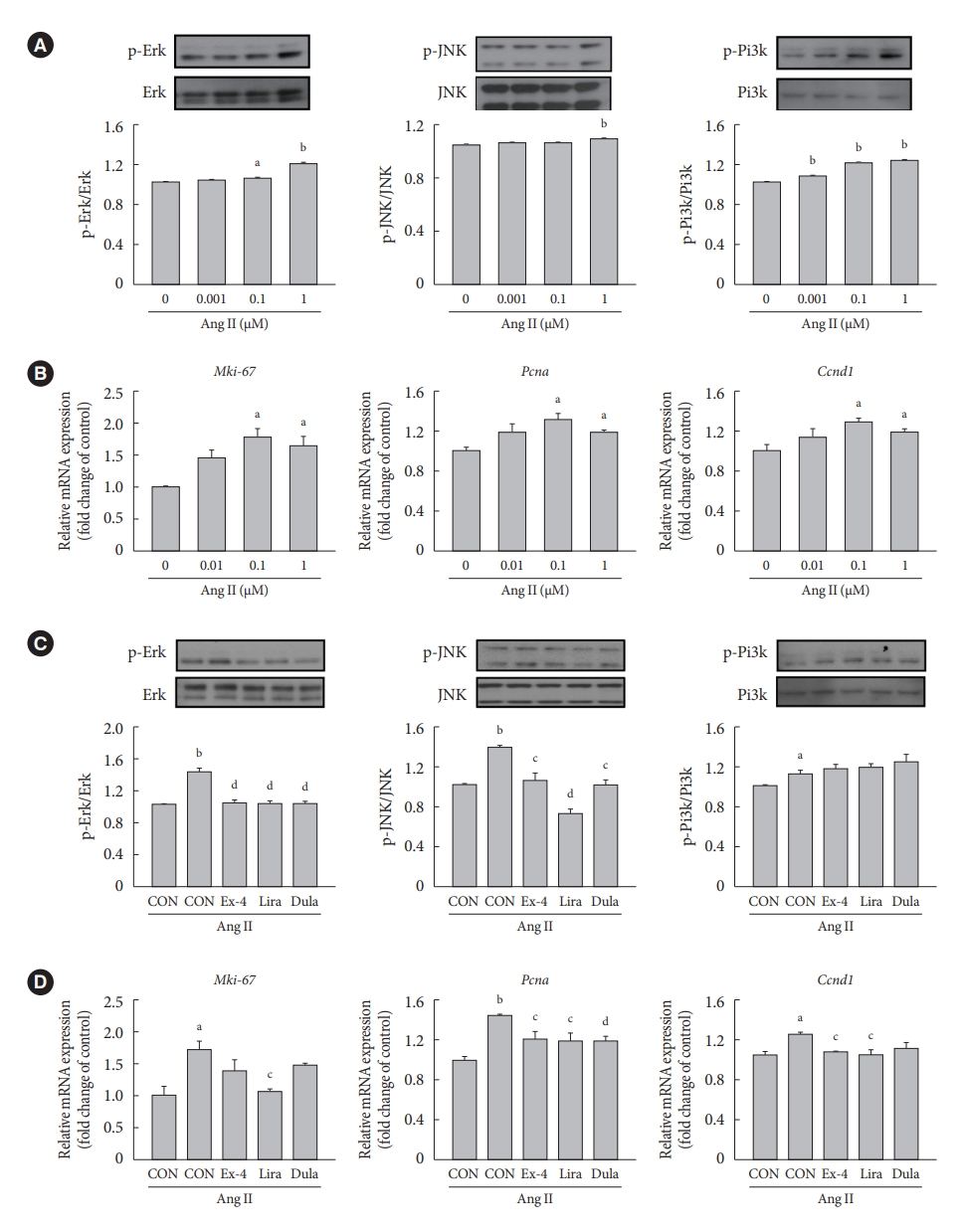
- GLP-1RAs decrease Erk and JNK activation and proliferation marker expression in Ang II-treated VSMCs
- We examined whether exendin-4, liraglutide, and dulaglutide regulate the expression of extracellular signal-regulated kinase (Erk), c-JUN N-terminal kinase (JNK), and phosphatidylinositol 3-kinase (Pi3k)/protein kinase B (Akt) signaling pathways in Ang II-treated cells. Ang II increased the protein expression of phospho-Erk (p-Erk), p-JNK, and p-Pi3k in a dose-dependent manner (Fig. 2A). Compared with controls, gene expression of marker of proliferation Ki-67 (Mki-67), Pcna, and cyclin D1 (Ccnd1) was increased the most in cells treated with 0.1 µM Ang II (Fig. 2B). Treatment with exendin-4, liraglutide, and dulaglutide decreased the expression of p-Ekr and p-JNK proteins (Fig. 2C) and Pcna gene (Fig. 2D) relative to control cells treated with Ang II alone. Also, GLP-1RAs decreased the expression of nuclear factor-κB (NF-κB), which is a downstream factor of Erk and JNK signaling pathways (Supplementary Fig. 2). Mki-67 expression was significantly decreased only in the cells treated with liraglutide, and Ccnd1 expression was decreased in the cells treated with exendin-4 and liraglutide compared with Ccnd1 expression in cells treated with Ang II alone. No significant difference was observed in the expression of p-Pi3k protein between cells treated with Ang II alone and those treated with exendin-4, liraglutide, or dulaglutide. Furthermore, immunofluorescence staining of Pcna revealed that the number of positive cells was lower among cells treated with exendin-4, liraglutide, or dulaglutide than among cells treated with Ang II alone (Supplementary Fig. 3).
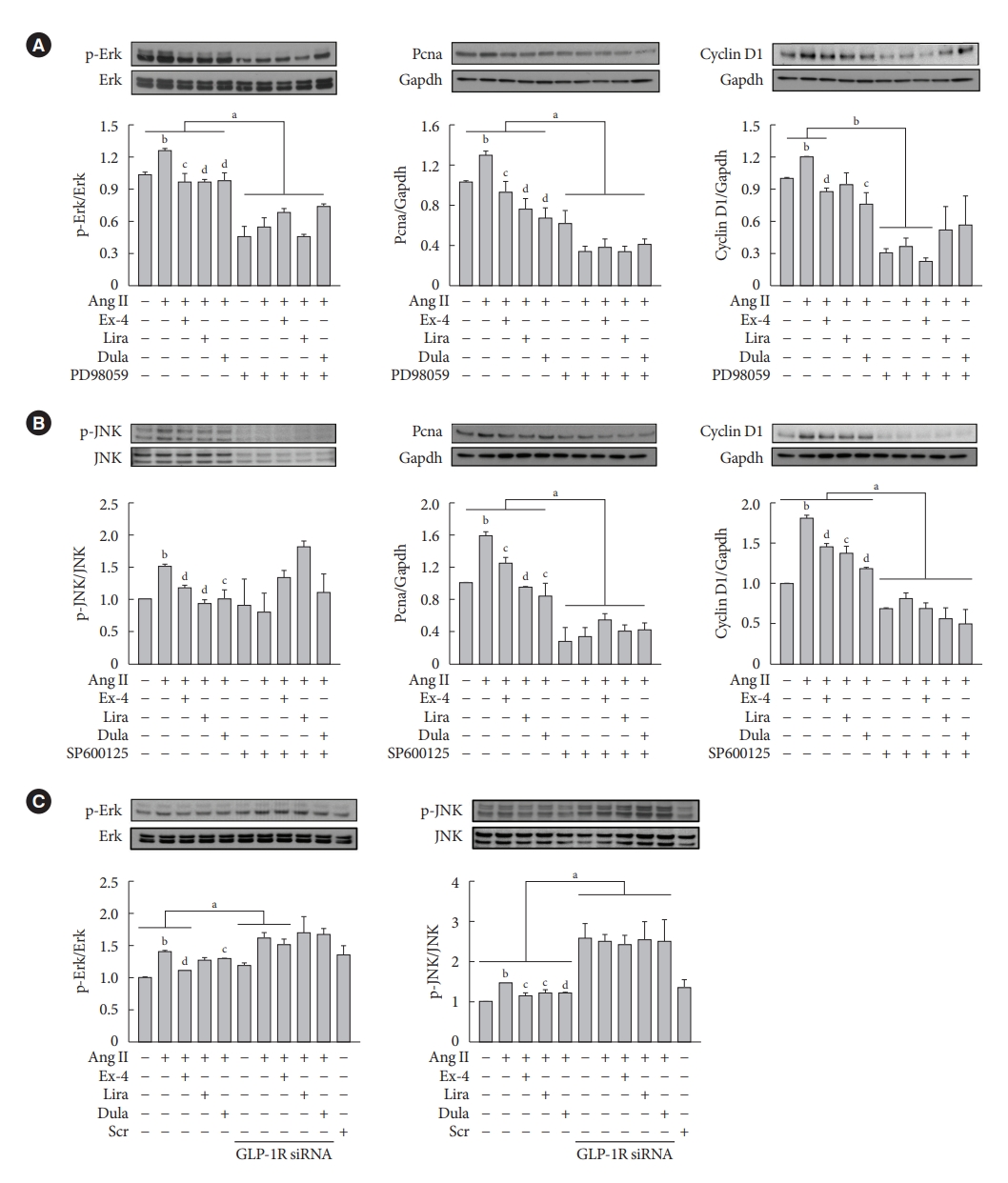
- GLP-1RAs decrease proliferation marker expression by inhibiting Erk and JNK in Ang II-treated VSMCs, and their effects are mediated by GLP-1R
- To determine whether the GLP-1RAs inhibits VSMC proliferation via Erk and JNK signaling pathways, cells were treated with PD98059 (Erk inhibitor) or SP600125 (JNK inhibitor). Inhibition of Ekr by PD98059 treatment decreased the expression of p-Erk, Pcna, and Ccnd1 proteins (Fig. 3A), while inhibition of JNK by SP600125 treatment decreased the expression of p-JNK, Pcna, and Ccnd1 proteins (Fig. 3B). Consistently, inhibition of Erk and JNK by inhibitor treatment significantly reduced cell proliferation compared with inhibitor-untreated groups (Supplementary Fig. 4). While lipopolysaccharide-induced Erk and JNK activation reversed the reduction by GLP-1RAs in VSMC migration (Supplementary Fig. 5). Next, we examined whether the inhibition of Erk and JNK signaling pathway by exendin-4, liraglutide, and dulaglutide treatment in Ang II-treated VSMCs was mediated through a GLP-1R-dependent pathway. Cells were transfected with GLP-1R siRNA, and we observed that GLP-1R inhibition by siRNA reversed the reduction of p-Erk and p-JNK expression by GLP-1RAs. (Fig. 3C). These data show that inhibitory effects by exendin-4, liraglutide, and dulaglutide in proliferation and migration of VSMCs are mediated through a GLP-1R-dependent pathway.
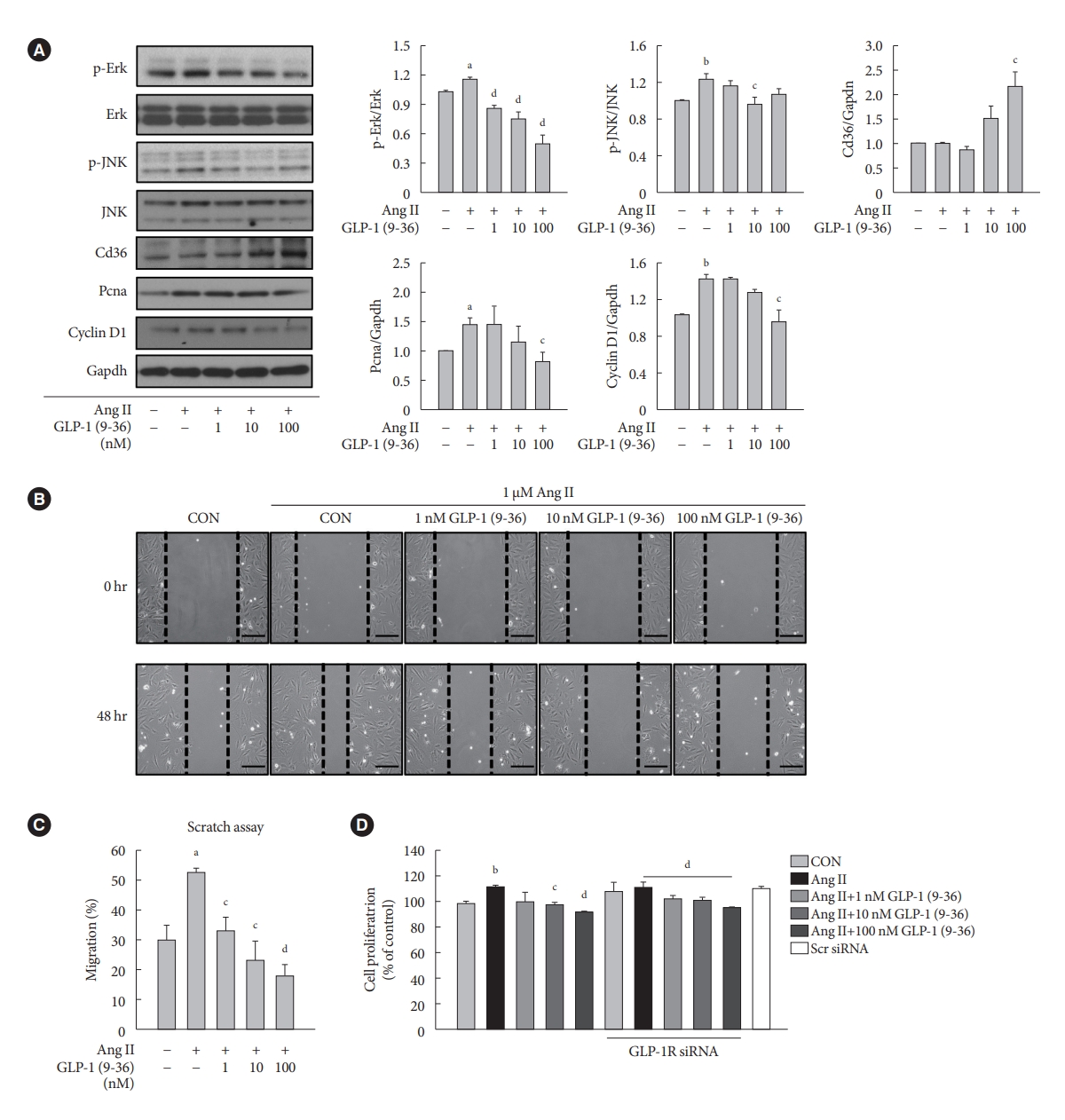
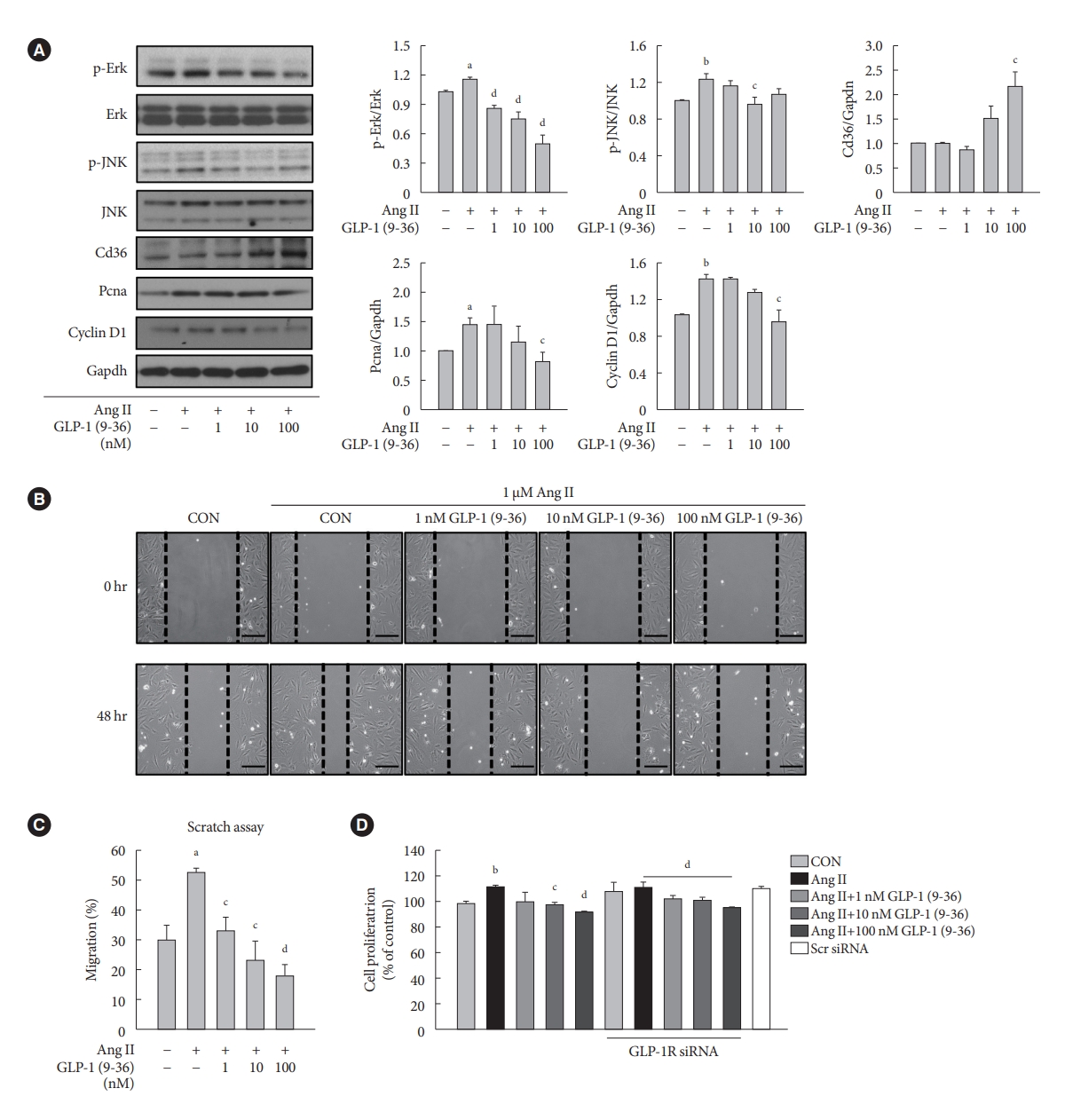
- GLP-1 (9-36) amide decreases proliferation and migration in Ang II-treated VSMCs and increases the expression of Cd36
- To demonstrate the involvement of the GLP-1R-independent pathway in the cardioprotective effect of GLP-1, Ang II-treated VSMCs were exposed to various concentrations of GLP-1 (9-36) amide, which is a metabolite of intact GLP-1 (7-36). GLP-1 (9-36) amide increased the protein expression of Cd36, a multifunctional membrane signaling receptor, and decreased the protein expression of p-Erk, p-JNK, Pcna, and Ccnd1 (Fig. 4A). In addition, GLP-1 (9-36) amide inhibited the migration of Ang II-induced VSMCs (Fig. 4B and C). Interestingly, the anti-proliferative effect in cells treated with 100 nM GLP-1(9-36) was also observed in cells transfected with GLP-1R siRNA (Fig. 4D). These findings indicate that GLP-1 (9-36) amide inhibits the aberrant migration and proliferation of VSMCs through a GLP-1R independent pathway.
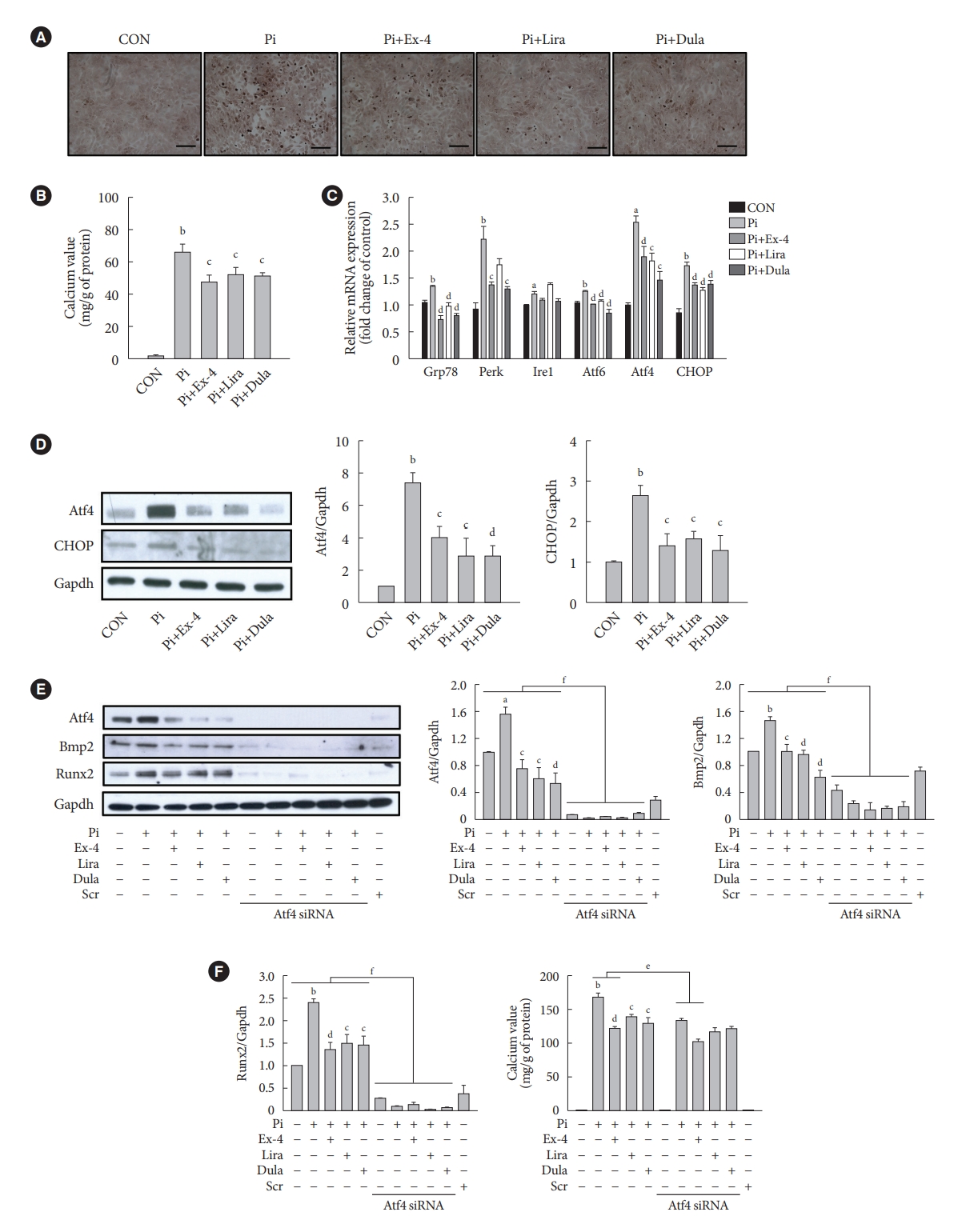
- GLP-1RAs decrease Pi-induced calcium deposition by inhibiting Atf4 in VSMCs
- We examined the effect of exendin-4, liraglutide, and dulaglutide on Pi-induced vascular calcification in VSMCs. GLP-1RAs diminished calcium deposition compared with that in the cells treated by Pi alone (Fig. 5A and B). The gene expressions of ER stress markers 78 kDa glucose-regulated protein (Grp78), protein kinase RNA-like endoplasmic reticulum kinase (Perk), inositol-requiring protein 1 (Ire1), activating transcription factor 6 (Atf6), Atf4, and C/EBP homologous protein (CHOP) and the protein expression of Atf4 transcription factor and CHOP, which are downstream factors of Perk, were increased in the Pi-treated cells compared with that in the control group, while it was reversed in cells treated with GLP-1RAs (Fig. 5C and D). In addition, the inhibition of Atf4 by siRNA dramatically decreased the expression of bone morphogenic protein 2 (Bmp2) and runt-related transcription factor-2 (Runx2) proteins, which are markers of osteoblastic differentiation in VSMCs (Fig. 5E), and diminished calcium deposition in cells treated Pi alone and Pi+exendin-4 (Fig. 5F). While ER stress inducer thapsigargin-induced Atf4 activation reversed the reduced calcium deposition by GLP-1RAs (Supplementary Fig. 6). These data indicate that GLP-1RAs have anti-calcification effects in Pi-treated VSMCs, and beneficial effects of GLP-1RAs are mediated through inhibition of osteoblastic differentiation of VSMCs via Atf4 inhibition.
RESULTS
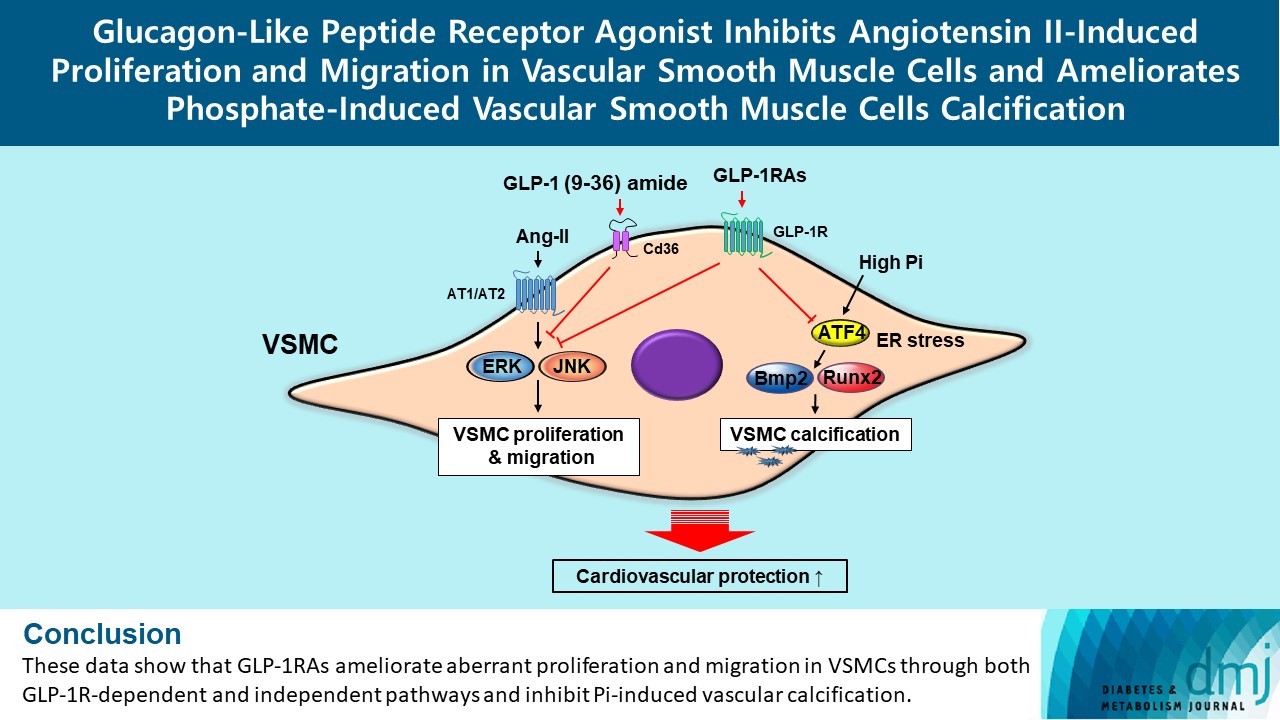
- In this study, we determined that GLP-1RAs have protective effects against the progression of coronary atherosclerosis via both GLP-1R-dependent and GLP-1R-independent pathways. GLP-1RA also has an anti-calcification effect by inhibition of Atf4. The GLP-1RAs exendin-4, liraglutide, and dulaglutide inhibited the aberrant increase in migration and proliferation of Ang II-treated VSMCs and decreased the expression of proliferation markers Pcna and Ccnd1 by inhibiting the Erk and JNK signaling pathways, which are involved in processes of cell growth and survival. However, GLP-1R inhibition by siRNA reversed the reduction of p-Erk and p-JNK expression caused by GLP-1RAs. Moreover, GLP-1 (9-36) amide showed similar improvement effects as GLP-1RAs on the proliferation and migration of VSMCs and the anti-proliferative effect of GLP-1 (9-36) amide is mediated by a GLP-1R-independent pathway. Furthermore, in rat VSMCs treated with a high concentration of Pi, exendin-4, liraglutide, and dulaglutide inhibited calcium deposition and decreased the expression of osteoblastic differentiation markers Bmp2 and Runx2 by inhibiting Atf4 (Fig. 6).
- The extraglycemic effect of the anti-diabetic medication is becoming increasingly important for the prevention of diabetes complications. Recently, a number of studies reported the results of CVD trials investigating the effect of GLP-1RAs including lixisenatide, liraglutide, semaglutide, exenatide, albiglutide, dulaglutide, and oral semaglutide in patients with T2DM, and established CVD risk factors [29,30]. A meta-analysis pooling the results of these randomized controlled trials showed that GLP-1RA treatment significantly reduced the risk of major adverse cardiac events by 12%, death due to CVD by 11%, stroke by 16%, myocardial infarction by 9%, and allcause death by 12% compared with a placebo group [31].
- GLP-1 is a gut hormone that has a beneficial effect on non-alcoholic fatty liver disease and CVD in patients with T2DM, through GLP-1R [32]. GLP-1 (9-36) amide, which is a metabolite of GLP-1 produced by DPP-4 cleavage, is metabolically inactive [33]. However, some previous studies reported that protective functions of GLP-1, exendin-4, and GLP-1 (9-36) amide against ischemia-reperfusion injury in hearts isolated from mice and rats [27,34]. The insulinomimetic effects of GLP-1 derived peptides, including GLP-1 (9-36) amide and GLP-1 (28-36) amide, modulated hepatic glucose production, antioxidant cardioprotective action, oxidative stress in vasculature tissue [35], and improved β-cell injury in the streptozotocindiabetic mice [36]. In this study, we observed that exendin-4, liraglutide, and dulaglutide inhibited the proliferation and migration of Ang II-treated VSMCs and reduced the expression of proliferation markers, Pcna and Ccnd1 by inhibiting Erk and JNK signaling via the GLP-1R-dependent pathway. In addition, GLP-1 (9-36) amide caused a dose-dependent decrease in VSMC migration and proliferation. Tomas and Habener [35] reported that GLP-1 (9-36) amide is transported in the cell through binding to CD36/fatty acid translocase, which is a receptor involved in the transport of peptides and oxidized LDLs. Our results show that GLP-1 (9-36) amide increased the expression of Cd36 in Ang II-treated VSMCs in a dose-dependent manner. These results indicate that GLP-1RAs have direct protective effects against vascular disease through GLP-1R-dependent and GLP-1R-independent pathways.
- Vascular calcification is a marker of CVD. In particular, calcification in the medial layer of the artery wall, which is composed of VSMCs, is closely related to diabetes [37]. VSMCs treated with a high concentration of Pi results in vascular calcification through various mechanisms including transdifferentiation of VSMCs into osteoblast-like cells, apoptosis, mineral vesicle release, ER stress, and oxidative stress [38,39]. We observed that exendin-4, liraglutide, and dulaglutide inhibited calcium deposition and reduced the gene expression of ER stress markers in Pi-treated VSMCs. Furthermore, we observed that Atf4 inhibition reduced the expression of osteoblastic differentiation markers Bmp2 and Runx2 and calcium deposition in Pi-treated VSMCs. These results indicate that GLP-1RAs have protective effects against vascular calcification in Pi-treated VSMCs, and these effects could be mediated by inhibition of VSMCs transdifferentiation into osteoblast-like cells via amelioration of ER stress.
- In conclusion, the present study showed that GLP-1RAs have a direct protective effect against vascular disease, and their anti-atherosclerotic effects might be mediated by both GLP-1R-dependent and GLP-1R-independent pathways. The GLP-1RAs treatment in Ang II-treated VSMCs inhibited migration and proliferation of VSMCs by inhibiting the Erk and JNK signaling pathways. Moreover, GLP-1 (9-36) amide also showed an inhibitory effect on VSMC migration and proliferation. We also found that GLP-1RAs had anti-calcification effects via inhibition of osteoblastic differentiation in Pi-treated VSMCs, which was mediated by Atf4 inhibition. Thus, treatment with GLP-1RAs may be a useful approach in the treatment of atherosclerosis and vascular calcification, which are the causes and risk factors for CVD. Further studies are required to clarify the role and underlying regulatory mechanism of Cd36 on the migration and proliferation of VSMCs by GLP-1 (9-36) amide. In addition, further studies are required to elucidate which major receptors mediate the beneficial effects of GLP-1 (9-36) amide in the coronary arteries, and in vivo experiments are needed to solidify our findings.
DISCUSSION
SUPPLEMENTARY MATERIALS
Supplementary Table 1.
Supplementary Table 2.
Supplementary Fig. 1.
Supplementary Fig. 2.
Supplementary Fig. 3.
Supplementary Fig. 4.
Supplementary Fig. 5.
Supplementary Fig. 6.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conception or design: J.L., S.W.H., S.J.M., E.J.R., W.Y.L.
Acquisition, analysis, or interpretation of data: all authors.
Drafting the work or revising: all authors.
Final approval of the manuscript: J.L., S.W.H., E.J.R., W.Y.L.
-
FUNDING
This study was supported by the National Research Foundation (NRF), which is funded by the Korean government (NRF2020R1F1A1073374) (http://www.nrf.re.kr), and was supported by Young Medical Scientist Research Grant through the Daewoong Foundation (DY20105P). The funders had no role in the study design, data collection, analysis, the decision to publish, or preparation of the manuscript.
NOTES
-
Acknowledgements
- None

- 1. Anderson SL, Marrs JC. Antihyperglycemic medications and cardiovascular risk reduction. Eur Endocrinol 2017;13:86-90.ArticlePubMedPMC
- 2. Pozo L, Bello F, Suarez A, Ochoa-Martinez FE, Mendez Y, Chang CH, et al. Novel pharmacological therapy in type 2 diabetes mellitus with established cardiovascular disease: current evidence. World J Diabetes 2019;10:291-303.ArticlePubMedPMC
- 3. Naito R, Miyauchi K. Coronary artery disease and type 2 diabetes mellitus. Int Heart J 2017;58:475-80.ArticlePubMed
- 4. Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 2019;16:727-44.ArticlePubMedPDF
- 5. van Diepen JA, Berbee JF, Havekes LM, Rensen PC. Interactions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis 2013;228:306-15.ArticlePubMed
- 6. Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, et al. Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol 2019;39:876-87.ArticlePubMedPMC
- 7. Grootaert MOJ, Bennett MR. Vascular smooth muscle cells in atherosclerosis: time for a re-assessment. Cardiovasc Res 2021;117:2326-39.ArticlePubMedPMCPDF
- 8. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016;118:692-702.ArticlePubMedPMC
- 9. van Thiel BS, van der Pluijm I, te Riet L, Essers J, Danser AH. The renin-angiotensin system and its involvement in vascular disease. Eur J Pharmacol 2015;763(Pt A):3-14.ArticlePubMed
- 10. Dasgupta C, Zhang L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov Today 2011;16:22-34.ArticlePubMed
- 11. Lemarie CA, Schiffrin EL. The angiotensin II type 2 receptor in cardiovascular disease. J Renin Angiotensin Aldosterone Syst 2010;11:19-31.ArticlePubMedPDF
- 12. Yang J, Chen C, Ren H, Han Y, He D, Zhou L, et al. Angiotensin II AT(2) receptor decreases AT(1) receptor expression and function via nitric oxide/cGMP/Sp1 in renal proximal tubule cells from Wistar-Kyoto rats. J Hypertens 2012;30:1176-84.ArticlePubMedPMC
- 13. Guimond MO, Gallo-Payet N. How does angiotensin AT(2) receptor activation help neuronal differentiation and improve neuronal pathological situations? Front Endocrinol (Lausanne) 2012;3:164.ArticlePubMedPMC
- 14. Chai W, Wang W, Dong Z, Cao W, Liu Z. Angiotensin II receptors modulate muscle microvascular and metabolic responses to insulin in vivo. Diabetes 2011;60:2939-46.ArticlePubMedPMCPDF
- 15. Zhao Y, Shang F, Shi W, Zhang J, Zhang J, Liu X, et al. Angiotensin II receptor type 1 antagonists modulate vascular smooth muscle cell proliferation and migration via AMPK/mTOR. Cardiology 2019;143:1-10.ArticlePubMedPDF
- 16. Trion A, van der Laarse A. Vascular smooth muscle cells and calcification in atherosclerosis. Am Heart J 2004;147:808-14.ArticlePubMed
- 17. Shi X, Gao J, Lv Q, Cai H, Wang F, Ye R, et al. Calcification in atherosclerotic plaque vulnerability: friend or foe? Front Physiol 2020;11:56.ArticlePubMedPMC
- 18. Proudfoot D, Shanahan CM. Biology of calcification in vascular cells: intima versus media. Herz 2001;26:245-51.ArticlePubMedPDF
- 19. Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J 2014;35:1515-25.ArticlePubMedPMC
- 20. Lau WL, Festing MH, Giachelli CM. Phosphate and vascular calcification: emerging role of the sodium-dependent phosphate co-transporter PiT-1. Thromb Haemost 2010;104:464-70.ArticlePubMedPMC
- 21. Saraiva FK, Sposito AC. Cardiovascular effects of glucagon-like peptide 1 (GLP-1) receptor agonists. Cardiovasc Diabetol 2014;13:142.ArticlePubMedPMCPDF
- 22. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311-22.ArticlePubMedPMC
- 23. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016;375:1834-44.ArticlePubMed
- 24. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995;136:3585-96.ArticlePubMed
- 25. Meier JJ, Gethmann A, Nauck MA, Gotze O, Schmitz F, Deacon CF, et al. The glucagon-like peptide-1 metabolite GLP-1-(9-36) amide reduces postprandial glycemia independently of gastric emptying and insulin secretion in humans. Am J Physiol Endocrinol Metab 2006;290:E1118-23.ArticlePubMed
- 26. Li J, Zheng J, Wang S, Lau HK, Fathi A, Wang Q. Cardiovascular benefits of native GLP-1 and its metabolites: an indicator for GLP-1-therapy strategies. Front Physiol 2017;8:15.ArticlePubMedPMC
- 27. Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagonlike peptide 1 receptor are mediated through both glucagonlike peptide 1 receptor-dependent and -independent pathways. Circulation 2008;117:2340-50.ArticlePubMed
- 28. Shi L, Ji Y, Jiang X, Zhou L, Xu Y, Li Y, et al. Liraglutide attenuates high glucose-induced abnormal cell migration, proliferation, and apoptosis of vascular smooth muscle cells by activating the GLP-1 receptor, and inhibiting ERK1/2 and PI3K/Akt signaling pathways. Cardiovasc Diabetol 2015;14:18.ArticlePubMedPMCPDF
- 29. Sheahan KH, Wahlberg EA, Gilbert MP. An overview of GLP1 agonists and recent cardiovascular outcomes trials. Postgrad Med J 2020;96:156-61.ArticlePubMedPMCPDF
- 30. Giugliano D, Scappaticcio L, Longo M, Caruso P, Maiorino MI, Bellastella G, et al. GLP-1 receptor agonists and cardiorenal outcomes in type 2 diabetes: an updated meta-analysis of eight CVOTs. Cardiovasc Diabetol 2021;20:189.ArticlePubMedPMCPDF
- 31. Rhee EJ. Extra-glycemic effects of anti-diabetic medications: two birds with one stone? Endocrinol Metab (Seoul) 2022;37:415-29.ArticlePubMedPMCPDF
- 32. Caussy C, Aubin A, Loomba R. The relationship between type 2 diabetes, NAFLD, and cardiovascular risk. Curr Diab Rep 2021;21:15.ArticlePubMedPMCPDF
- 33. Knudsen LB, Pridal L. Glucagon-like peptide-1-(9-36) amide is a major metabolite of glucagon-like peptide-1-(7-36) amide after in vivo administration to dogs, and it acts as an antagonist on the pancreatic receptor. Eur J Pharmacol 1996;318:429-35.ArticlePubMed
- 34. Sonne DP, Engstrom T, Treiman M. Protective effects of GLP-1 analogues exendin-4 and GLP-1(9-36) amide against ischemia-reperfusion injury in rat heart. Regul Pept 2008;146:243-9.ArticlePubMed
- 35. Tomas E, Habener JF. Insulin-like actions of glucagon-like peptide-1: a dual receptor hypothesis. Trends Endocrinol Metab 2010;21:59-67.ArticlePubMedPMC
- 36. Shao W, Wang Z, Ip W, Chiang YT, Xiong X, Chai T, et al. GLP1(28-36) improves β-cell mass and glucose disposal in streptozotocin-induced diabetic mice and activates cAMP/PKA/ β-catenin signaling in β-cells in vitro. Am J Physiol Endocrinol Metab 2013;304:E1263-72.ArticlePubMed
- 37. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Mechanisms of medial arterial calcification in diabetes. Curr Pharm Des 2014;20:5870-83.ArticlePubMed
- 38. Lee J, Hong SW, Kim MJ, Kwon H, Park SE, Rhee EJ, et al. Metformin, resveratrol, and exendin-4 inhibit high phosphate-induced vascular calcification via AMPK-RANKL signaling. Biochem Biophys Res Commun 2020;530:374-80.ArticlePubMed
- 39. Al-Aly Z. Phosphate, oxidative stress, and nuclear factor-κB activation in vascular calcification. Kidney Int 2011;79:1044-7.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Incretin Hormone Secretion in Women with Polycystic Ovary Syndrome: Roles of Obesity, Insulin Sensitivity and Treatment with Metformin and GLP-1s
Andrea Etrusco, Mislav Mikuš, Antonio D’Amato, Fabio Barra, Petar Planinić, Trpimir Goluža, Giovanni Buzzaccarini, Jelena Marušić, Mara Tešanović, Antonio Simone Laganà
Biomedicines.2024; 12(3): 653. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite