- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 35(5); 2011 > Article
-
ReviewFuel-Stimulated Insulin Secretion Depends upon Mitochondria Activation and the Integration of Mitochondrial and Cytosolic Substrate Cycles
- Gary W. Cline
-
Diabetes & Metabolism Journal 2011;35(5):458-465.
DOI: https://doi.org/10.4093/dmj.2011.35.5.458
Published online: October 31, 2011
Department of Internal Medicine, Yale University School of Medicine, New Haven, CT, USA.
- Corresponding author: Gary W. Cline. Department of Internal Medicine, Yale University School of Medicine, TAC 269B 300 Cedar Street, New Haven, CT 06510, USA. gary.cline@yale.edu
Copyright © 2011 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- UNCOUPLING AS A REGULATOR OF MITOCHONDRIAL ACTIVATION: UCP2 ACTIVATION AND H+ DEPENDENT MITOCHONDRIAL CARRIERS
- MITOCHONDRIAL METABOLISM IS ACTIVATED BY MATRIX ALKALINIZATION
- ANAPLEROSIS AND MITOCHONDRIAL-CYTOSOLIC PYRUVATE CYCLING
- OTHER NOVEL MITOCHONDRIAL SUBSTRATE CYCLES
- RELEVANCE TO HUMAN ISLET BIOLOLGY
- ARE OUR MECHANISTIC MODELS RELEVANT TO GSIS IN VIVO?
- ACKNOWLEDGMENTS
- NOTES
- REFERENCES
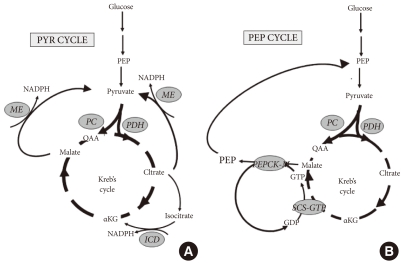
Figure & Data
References
Citations
Citations to this article as recorded by 
- Nicotinamide Adenine Dinucleotide: The Redox Sensor in Aging-Related Disorders
Giovanna Schiuma, Djidjell Lara, James Clement, Marco Narducci, Roberta Rizzo
Antioxidants & Redox Signaling.2024;[Epub] CrossRef - ROCK1 regulates insulin secretion from β-cells
Byung-Jun Sung, Sung-Bin Lim, Won-Mo Yang, Jae Hyeon Kim, Rohit N. Kulkarni, Young-Bum Kim, Moon-Kyu Lee
Molecular Metabolism.2022; 66: 101625. CrossRef - Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity
Xiaoting Luo, Jinzi Wu, Siqun Jing, Liang-Jun Yan
Aging and disease.2016; 7(1): 90. CrossRef - Characterization of Phospholipids in Insulin Secretory Granules and Mitochondria in Pancreatic Beta Cells and Their Changes with Glucose Stimulation
Michael J. MacDonald, Lacmbouh Ade, James M. Ntambi, Israr-Ul H. Ansari, Scott W. Stoker
Journal of Biological Chemistry.2015; 290(17): 11075. CrossRef - Roles of Pyruvate, NADH, and Mitochondrial Complex I in Redox Balance and Imbalance inβCell Function and Dysfunction
Xiaoting Luo, Rongrong Li, Liang-Jun Yan
Journal of Diabetes Research.2015; 2015: 1. CrossRef - Peroxisome Proliferator-activated Receptor γ (PPARγ) and Its Target Genes Are Downstream Effectors of FoxO1 Protein in Islet β-Cells
Dhananjay Gupta, Averi A. Leahy, Navjot Monga, Mina Peshavaria, Thomas L. Jetton, Jack L. Leahy
Journal of Biological Chemistry.2013; 288(35): 25440. CrossRef - Desnutrin/ATGL Activates PPARδ to Promote Mitochondrial Function for Insulin Secretion in Islet β Cells
Tianyi Tang, Marcia J. Abbott, Maryam Ahmadian, Andressa B. Lopes, Yuhui Wang, Hei Sook Sul
Cell Metabolism.2013; 18(6): 883. CrossRef - Potent humanin analog increases glucose‐stimulated insulin secretion through enhanced metabolism in the β cell
Regina Kuliawat, Laura Klein, Zhenwei Gong, Marianna Nicoletta‐Gentile, Anjana Nemkal, Lingguang Cui, Claire Bastie, Kai Su, Derek Huffman, Manju Surana, Nir Barzilai, Norman Fleischer, Radhika Muzumdar
The FASEB Journal.2013; 27(12): 4890. CrossRef - Proliferation and differentiation of osteoblasts from the mandible of osteoporotic rats
Shu-Juan Yu, Hong-Chen Liu, Ling-Ling E, Dong-Sheng Wang, Guo-Xiong Zhu
Experimental Biology and Medicine.2012; 237(4): 395. CrossRef
 PubReader
PubReader Cite
Cite