- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 46(1); 2022 > Article
-
ReviewPathophysiology Insulin Resistance: From Mechanisms to Therapeutic Strategies
-
Shin-Hae Lee1
 , Shi-Young Park1, Cheol Soo Choi1,2,3
, Shi-Young Park1, Cheol Soo Choi1,2,3 -
Diabetes & Metabolism Journal 2022;46(1):15-37.
DOI: https://doi.org/10.4093/dmj.2021.0280
Published online: December 30, 2021
1Korea Mouse Metabolic Phenotyping Center (KMMPC), Lee Gil Ya Cancer and Diabetes Institute, Gachon University, Incheon, Korea
2Department of Internal Medicine, Gachon University Gil Medical Center, Incheon, Korea
3Division of Molecular Medicine, Gachon University College of Medicine, Incheon, Korea
- Corresponding author: Cheol Soo Choi https://orcid.org/0000-0001-9627-058X Division of Molecular Medicine, Gachon University College of Medicine, 21 Namdongdaero 774beon-gil, Namdong-gu, Incheon 21565, Korea E-mail: cschoi@gachon.ac.kr
Copyright © 2022 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Insulin resistance is the pivotal pathogenic component of many metabolic diseases, including type 2 diabetes mellitus, and is defined as a state of reduced responsiveness of insulin-targeting tissues to physiological levels of insulin. Although the underlying mechanism of insulin resistance is not fully understood, several credible theories have been proposed. In this review, we summarize the functions of insulin in glucose metabolism in typical metabolic tissues and describe the mechanisms proposed to underlie insulin resistance, that is, ectopic lipid accumulation in liver and skeletal muscle, endoplasmic reticulum stress, and inflammation. In addition, we suggest potential therapeutic strategies for addressing insulin resistance.
- Evolution has equipped animals with highly efficient means of overcoming nutrient scarcity, which involves the synthesis of complex molecules and storage of redeemable energy sources when nutrients are plentiful. Insulin is a pivotal regulator of the transition from nutrient production to storage under such conditions. After nutrient intake, plasma glucose levels reach a threshold level that stimulates insulin secretion by pancreatic β-cells. Under normal circumstances, this insulin promotes carbohydrate uptake at key storage and consumption sites, such as in adipose tissue and skeletal muscle, in which carbohydrates and proteins are stored as lipids. However, over-nourished, sedentary modern lifestyles disrupt this system and can cause serious medical problems, which include metabolic syndrome, obesity, type 2 diabetes mellitus (T2DM), and cardiovascular disease.
- Insulin resistance is defined physiologically as a state of reduced responsiveness in insulin-targeting tissues to high physiological insulin levels and is considered the pathogenic driver of many modern diseases, including metabolic syndrome, nonalcoholic fatty liver disease (NAFLD), atherosclerosis, and T2DM [1]. Insulin resistance precedes non-physiologic elevated plasma glucose levels, which is the primary clinical symptom of T2DM. In the prediabetic condition, insulin levels increase to meet normal insulin requirements leading to chronic hyperinsulinemia, hyperglycemia-induced β-cell failure, and eventually to T2DM [2].
- Although the mechanism of insulin resistance has not been fully established, several theories are generally considered reasonable. This review summarizes the function of insulin in glucose metabolism in metabolic tissues, such as liver, skeletal muscle, and adipose tissue, and describes several putative mechanisms of insulin resistance, including the ectopic accumulation of lipids in liver and skeletal muscle. In addition, we suggest potential therapeutic strategies for fat-induced insulin resistance that target ectopic fat accumulation in liver and promote energy consumption by skeletal muscle.
INTRODUCTION
- Insulin signaling
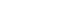
- In the fasted state, the liver secretes glucose into blood to maintain euglycemia and provide fuel for glucose-consuming tissues. This process is called hepatic glucose production (HGP), and involves the breakdown of hepatic glycogen (glycogenolysis) and de novo synthesis of glucose (gluconeogenesis) using fatty acids and glycerol derived from adipose tissues [3]. After food intake, insulin secreted by pancreatic β-cells promotes anabolism and suppresses catabolic programs. During glucose metabolism, insulin stimulates several glucose-consuming tissues, such as skeletal muscle and adipose tissues, to uptake glucose and then promotes the syntheses of glycogen and lipid in liver, skeletal muscle, and adipose tissue [4]. In addition, insulin suppresses HGP by inhibiting the expressions of gluconeogenic genes and lipolysis in adipose tissue [5]. Insulin also suppresses glucagon secretion from pancreatic α-cells [6,7] and reduces appetite via the central nervous system. In this review, we focus on the role of insulin in glucose metabolism in skeletal muscle, liver, and adipose tissue.
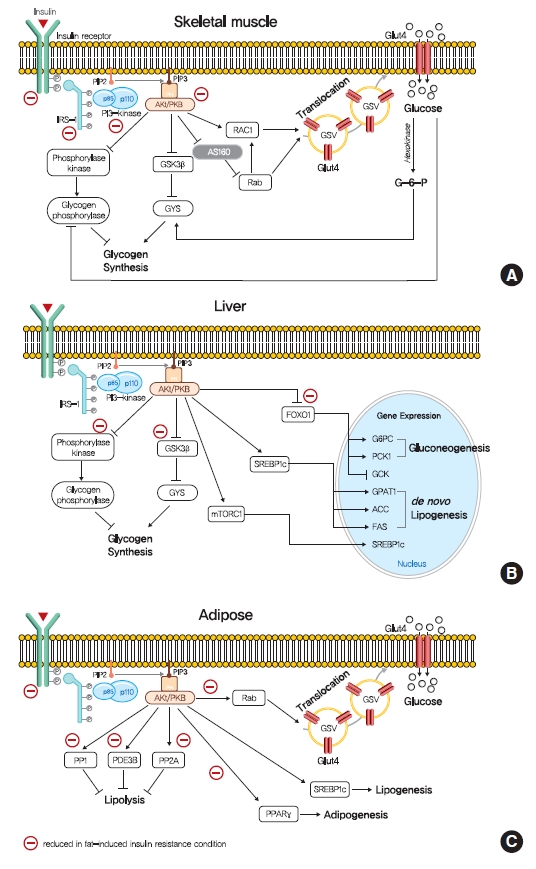
- The intracellular functions of insulin are mediated through insulin receptor tyrosine kinase (IRTK) (Fig. 1). When insulin binds to the extracellular domain of IRTK, it induces a conformational change that results in the autophosphorylation of IRTK tyrosine residues and the subsequent activation of phosphotyrosine-binding proteins such as insulin receptor substrate (IRS), growth factor receptor-bound protein-2 (GRB-2), GRB-10, SHC-transforming protein (SHC), and SH2B adapter protein-2 (SH2B-2) [8]. The effects of insulin on glucose and lipid metabolism are mainly mediated by the IRTK-induced phosphorylation of IRS, which then recruits phosphatidylinositol-3-OH kinase (PI3K) and catalyzes the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PIP2). After being recruited to the plasma membrane by PIP3, Akt is activated (phosphorylated) by 3-phosphoinositide-dependent kinase-1 (PDK1) and mechanistic target of rapamycin complex 2 (mTORC2) [9] and then phosphorylates various downstream substrates in metabolic tissues, including skeletal muscle, liver, and adipose tissue, which elicit insulin-induced nutrient reservation in these tissues.
- In skeletal muscle, insulin signaling promotes glucose uptake and net glycogen synthesis (Fig. 1A). Insulin increases glucose transport activity via the highly coordinated translocation and fusion of glucose transporter type 4 (GLUT4) storage vesicles (GSVs) to the plasma membrane in skeletal muscle [10]. After being activated by insulin signaling, Akt inactivates AS160 (GTPase-activating protein [GAP] AKT substrate of 160 kDa, also known as TBC1D4), which activates small Rab GTPase protein switches that control vesicle trafficking [10]. Insulin-induced Akt also promotes the guanosine triphosphate (GTP)-bound form of Ras-related C3 botulinum toxin substrate 1 (RAC1), which promotes GLUT4 translocation by inducing cortical actin reorganization [11]. On the other hand, insulin also regulates net glycogen synthesis in skeletal muscle by suppressing glycogenolysis and promoting glycogen synthesis. Insulin signaling promotes the activity of glycogen synthase (GYS) via the phosphorylation of glycogen synthase kinase 3 (GSK3) by Akt [12] and the activation of protein phosphatase 1 (PP1) to promote the dephosphorylation of GYS [13]. In addition, insulin regulates glycogen phosphorylase activity via the dephosphorylation of phosphorylase kinase [14].
- Insulin in liver activates IRTK, which phosphorylates IRS1 and IRS2, and ultimately activates Akt2 [15], which decreases HGP, promotes glycogen synthesis, and transcriptionally activates lipogenesis (Fig. 1B). The primary function of hepatic insulin signaling is to decrease HGP by repressing gluconeogenesis mediated by the Akt-induced phosphorylation of forkhead box O1 (FOXO1) [15], which excludes FOXO1 from the nucleus, and thus, prevents the transcriptional activations of gluconeogenic gene expressions, such as glucose-6-phosphatase (G6PC) and phosphoenolpyruvate carboxylase (PEPCK) [15,16]. In addition to inhibiting gluconeogenic gene expressions, insulin suppresses hepatic gluconeogenesis by inhibiting adipocyte lipolysis, which reduces levels of gluconeogenesis substrates in liver [17]. Furthermore, besides suppressing HGP, insulin increases hepatic glycogen synthesis by regulating GYS (especially GYS2 in liver) and glycogen phosphorylase through GSK3 and PP1, as occurs in skeletal muscle [18]. Also, insulin activates lipid anabolism by upregulating sterol regulatory element-binding protein 1c (SREBP-1c; a master transcriptional regulator of hepatic de novo lipogenesis) and subsequently, enhancing the transcriptions of several lipogenic genes, including acetyl-CoA carboxylase 1 (ACC1), fatty acid synthase (FAS), and glycerol-3-phosphate acyltransferase 1 (GPAT1) [19,20].
- The most important physiological function of insulin in white adipocyte tissue is to suppress lipolysis, which in turn suppresses HGP by reducing gluconeogenic substrates (Fig. 1C) [21]. The mechanism responsible for insulin-induced lipolysis suppression is not fully understood, though it is believed to be mediated by phosphodiesterase 3B (PDE3B) through reduced cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) activity [22]. In addition, PP1 and protein phosphatase-2A (PP2A) appear to mediate PI3K-dependent insulin-induced lipolysis suppression through the dephosphorylations of lipolytic regulatory proteins [23,24]. Although insulin promotes glucose transport by signaling the phosphorylation of targets involved in vesicle tethering, docking, and fusion, its contribution to whole-body glucose disposal is comparatively minor [25]. Insulin also promotes lipogenesis in white adipose tissue by activating SREBP-1c, signaling the translocations of glucose or fatty acid transport proteins (FATPs), promoting fatty acid esterification [26], and stimulating adipogenesis through the transcription factor peroxisome proliferator-activated receptor-γ (PPARγ) [27].
- Insulin resistance
- As mentioned above, insulin resistance is physiologically defined as an inability of some type of tissues to respond to normal insulin levels, and thus, higher than normal levels of insulin are required to maintain the normal functions of insulin. Notably, the glucose-regulating effects of insulin such as the suppressions of HGP and lipolysis, cellular uptake of plasma glucose, and net glycogen synthesis are not observed in insulin resistant tissues at normal plasma levels [5]. Since skeletal muscle is a quantitatively central tissue for insulin-stimulated glucose disposal, and liver and adipose tissue are qualitatively the critical sites for glucose-induced insulin signaling, these tissues are considered central to the understanding of the mechanisms responsible for insulin resistance.
- Insulin-stimulated glucose consumption is largely conducted in skeletal muscle, and thus, muscular insulin resistance could affect whole-body metabolism [28]. Muscle-specific insulin resistance by the muscle-specific deletion of IRTK or GLUT4 increased the susceptibility to hepatic steatosis, and increase adiposity in animal models [29,30]. Many molecular studies have demonstrated that insulin-stimulated muscle glucose uptake is highly susceptible to insulin resistance attributed to impaired GLUT4 translocation. However, the translocation of GLUT4 to the plasma membrane and glucose transport in T2DM are stimulated by hypoxia or exercise via the AMP-activated protein kinase (AMPK)-mediated regulation of GSV translocation [31,32], which suggests that defects of glucose transport in insulin resistance are abnormalities in the insulin signaling pathway rather than defects in the transport system itself. Furthermore, insulin resistance in skeletal muscle may be caused by defects at the proximal level of insulin signaling, for example, in the activities of IRTK, IRS1, PI3K, and AKT (Fig. 1A). In addition, in obese mice and obese/diabetic humans, the tyrosine kinase activity of IRTK is diminished in skeletal muscle, which supports this suggestion [33]. In addition, IRS1 tyrosine phosphorylation and IRS1-associated PI3K activity were found to be diminished in insulin resistant skeletal muscle [34].
- The liver critically controls postprandial carbohydrate levels by suppressing HGP and stimulating the deposition of glucose as glycogen and is the primary source of glucose production during fasting [35]. In T2DM patients, insulin cannot regulate hepatic glycogen synthesis or glucose production, and increased hepatic gluconeogenesis is the primary cause of fasting hyperglycemia in T2DM [36,37]. Defective suppression of hepatic gluconeogenesis in insulin resistance is largely associated with lipolysis defects in adipose tissue and the de-suppression of FOXO1 transcription factor in liver [35]. Meanwhile, insulin resistance is also associated with defects in the insulin-induced stimulation of glycogen synthesis, for example, T2DM patients have lower fasting and postprandial hepatic glycogen content [38]. Furthermore, hepatic insulin resistance reduces the amplitude of hepatic glycogen metabolism induced by fasting and feeding [36].
- Intriguingly, not all insulin functions are less responsive in the presence of insulin resistance. Some insulin pathways in hyperinsulinemia are highly responsive to insulin, a phenomenon referred to as selective insulin resistance or pathway-selective insulin responsiveness [39]. In the case of hepatic insulin resistance, insulin fails to suppress HGP but stimulates lipogenesis, which results in hyperglycemia, hyperlipidemia, and hepatic steatosis [39]. The underlying mechanism of selective insulin resistance has not been established but several hypotheses have been suggested. One involves glucose disposal potential differences in the substrate specificities of AKT phosphorylation between gluconeogenesis and lipogenesis [40]. Akt Ser473 phosphorylation may activate some AKT substrates related to gluconeogenesis, such as FOXO, and these activations might be suppressed in a background of insulin resistance, while other Akt substrates, such as GSK3β and tuberous sclerosis complex 2 (TSC2), which requires AKT phosphorylation at Thr308, might not be disrupted [41,42]. Another possible mechanism of selective insulin resistance in liver involves the different intrinsic sensitivities of insulin-induced SERBP-1c activation and gluconeogenesis suppression, which suggests that these functions require specific insulin levels. Recent studies have shown that insulin-regulated hepatic lipogenesis, but not gluconeogenesis, is mediated by stabilizing pleckstrin homology domain leucine-rich repeat protein phosphatase-2 (PHLPP-2), which terminates insulin-stimulated Akt activity [43,44]. These reports show that a high-fat diet (HFD) decreases PHLPP-2 stability, which delayed Akt dephosphorylation and then induces prolonged Akt activation in late stage after insulin pulse [43,44]. Since elevated Akt activity in the early postprandial stage is required to reduce HGP and Akt activity in the late stage is sufficient to increase de novo lipogenesis but not HGP [40,43,44], unterminated prolonged Akt activity might enhance de novo lipogenesis but not suppress HGP. The other hypothesis posits that selective insulin resistance in liver is caused by insulin-independent lipogenesis. Lipogenesis induction by nutrients is also mediated by the carbohydrate response element-binding protein (ChREBP), PPARγ coactivator 1-β, and liver X receptor-mediated SREBP-1c [45-47], and these alternative pathways of lipogenesis have been shown to be activated by fructose and monosaccharides [48,49].
INSULIN SIGNALING AND INSULIN RESISTANCE
Insulin resistance in skeletal muscle
Insulin resistance in liver and adipose tissue
Selective insulin resistance
- Many epidemiological studies have concluded that obesity is a leading risk factor of T2DM, and reportedly, obese people are up to 80 times more likely to develop T2DM [50]. Furthermore, it has been shown an inverse correlation exists between plasma fatty acid concentration and insulin sensitivity in age and body mass index matched controls [51]. These results and others show that excess lipid content caused by obesity is a primary cause of insulin resistance and T2DM. Many theories have been proposed for the mechanism responsible for the development of insulin resistance due to excess lipid availability.
- The glucose-fatty acid cycle (the Randle cycle)
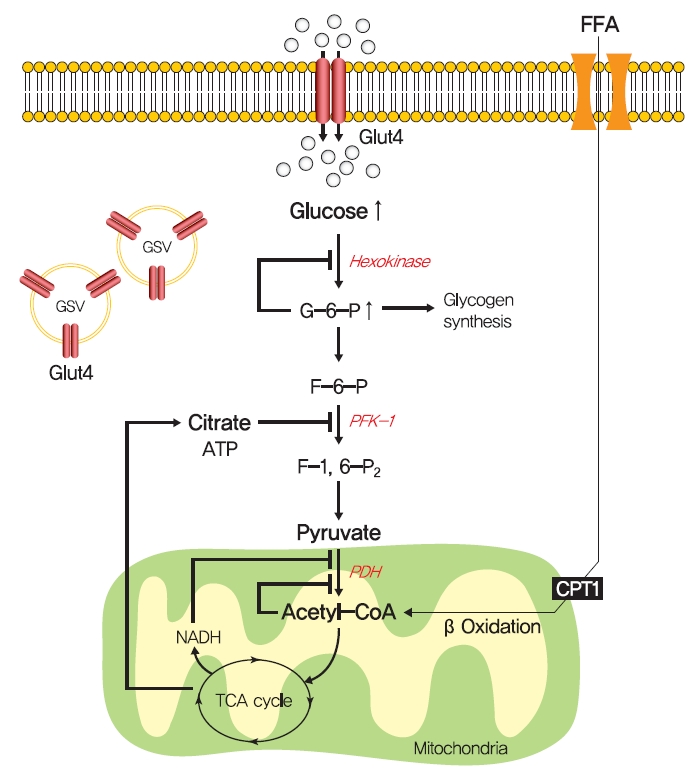
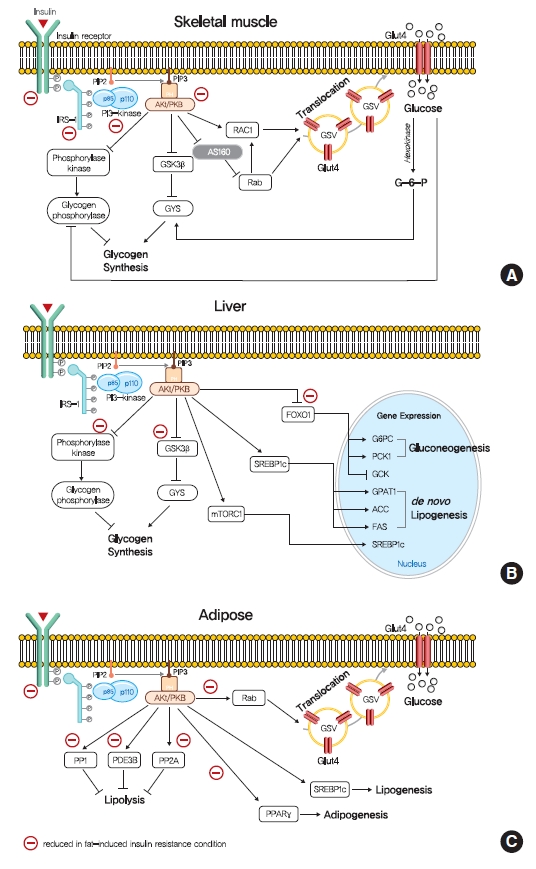
- In 1963, Randle et al. [52] hypothesized that lipid-induced insulin resistance in skeletal muscle characterized by a defect in glucose uptake is attributed to limited insulin-stimulated glucose utilization caused by increased fatty acid oxidation, which is referred to as the glucose-fatty acid or Randle cycle. It was asserted that increased fatty acid oxidation in the obese impaired in the use of glucose as a source of fuel by inhibiting the activities of key glycolytic enzymes. According to Randle et al. [52], fatty acid oxidation might increase mitochondrial acetyl-CoA levels through β-oxidation and subsequently inactivate pyruvate dehydrogenase, which in turn, would increase intracellular citrate levels and inhibit phosphofructokinase (a key glycolytic enzyme), and lead to the accumulation of intramyocellular glucose-6-phosphate, which inhibits hexokinase activity and causes the accumulation of intramyocellular glucose and reduced glucose uptake (Fig. 2). Furthermore, some experimental results have consistently shown that infusions of fatty acids decrease myocellular glucose utilization and increase intramyocellular glucose-6-phosphate concentrations in rat heart and diaphragm muscles [53-55].
- The glucose-fatty acid cycle shows that glucose-6-phosphate and glycogen synthesis levels should be increased by fatty acid and that glycolysis should be inhibited. However, exposure to high plasma fatty acid concentrations caused insulin resistance and reduced glucose-6-phosphate concentrations in the muscle of healthy individuals [56]. In addition, insulin-stimulated muscle glycogen synthesis and glucose oxidation were found to be suppressed in T2DM patients with chronic insulin resistance [57,58]. These studies demonstrate that the glucose-fatty acid cycle cannot fully explain all impairments of insulin resistance.
- Hexosamine biosynthesis pathway
- Although the glucose-fatty acid cycle does not satisfactorily explain lipid-induced insulin resistance, evidence indicates that muscular acetyl-CoA and citrate levels are elevated in line with increased fat oxidation, which suggests another pathway regulates glucose transport independently of glucose-6-phosphate levels in muscle (Fig. 3). The hexosamine biosynthesis pathway (HBP) offers another explanation for the development of insulin resistance. Fructose-6-phosphate is mainly produced from glucose-6-phosphate and is preferentially metabolized to fructose-1,6-bisphosphate during glycolysis, but approximately 5% of fructose-6-phosphate is converted to glucosamine-6-phosphate by glutamine:fructose-6-phosphate amidotransferase (GFAT), the rate-limiting enzyme of HBP [59]. Glucosamine-6-phosphate is converted to uridine 5’-diphosphate N-acetylglucosamine (UDP-GlcNAc), which serves as the donor sugar nucleotide for the glycosylation and O-GlcNAcylation of lipids and proteins [59], and these modifications, especially O-GlcNAcylation, could affect targeted proteins by regulating gene expressions or enzyme activities [60].
- Thirty years ago, Marshall et al. [61] first suggested O-GlcNAcylation modulated insulin sensitivity and showed that the effects of glucose-induced insulin resistance could be blocked by inhibiting GFAT using amidotransferase inhibitors such as O-diazoacetyl-L-serine (azaserine) or 6-diazo-5-oxonorleucine (DON). Subsequently, it was established that glucosamine induces insulin resistance [62,63]. Furthermore, hyperglycemia and hyperinsulinemia reportedly increase UDP-GlcNAc levels in skeletal muscle [64,65], and O-GlcNAc levels have been reported to be elevated in the corneas, pancreases, and skeletal muscles of diabetic or insulin resistant rats [66,67]. Also, GFAT overexpression in adipose tissue or skeletal muscle developed whole-body insulin resistance characterized by a reduction in glucose disposal in mice [68]. Recently, skeletal muscle-specific O-GlcNAc transferase (OGT) knockout mice on a HFD were reported to have low plasma glucose levels and glucose tolerances [69]. Moreover, the overexpression of O-GlcNAcase (OGA), which removes O-GlcNAc from proteins, significantly improved whole-body glucose tolerance and insulin sensitivity in db/db mice, and O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino-N-phenylcarbamate (PUGNAc) (an OGA inhibitor) suppressed insulin-mediated glucose uptake in adipocytes [70,71]. Also, single-nucleotide polymorphisms in meningioma-expressed antigen 5 (MGEA5), a human OGA homolog, were associated with T2DM onset in Pima Indians and Mexican Americans [72,73].
- The molecular mechanisms of HBP in insulin resistance have not been fully established, while O-GlcNAcylation provides the most acceptable mechanism of HBP in insulin resistance. O-GlcNAcylation of proteins has been suggested to compete with phosphorylation for sites, which could regulate the protein activity and signaling transduction [74]. In particular, insulin signaling pathway components, such as IRS-1/2, PI3K, PDK1, and Akt, are modified with O-GlcNAc [75,76]. In addition to these proximal components of insulin signaling, mammalian uncoordinated-18c (Munc18-c), a protein essential for insulin-stimulated GLUT4 translocation, was reported to be O-GlcNAc modified in a background of glucosamine-induced insulin resistance, and FOXO1, a master transcriptional factor for gluconeogenic genes in liver, was also O-GlcNacylated in diabetes [77,78].
- Debate continues despite cumulating evidence regarding the role of HBP in insulin resistance. Robinson et al. [79] showed that adenovirus-induced OGA overexpression and OGT knockdown failed to restore impaired Akt activation by insulin stimulation and prevent glucose-induced insulin resistance in 3T3L1 adipocytes, and in another study, treatment with the OGA inhibitor 1,2-dideoxy-2’-propyl-α-ᴅ-glucopyranoso-[2,1, D]-Δ2’-thiazoline (NButGT) did not induce insulin resistance characterized by defective glucose uptake and Akt phosphorylation in 3T3-L1 adipocytes [80]. Taken together, discrepancies between the roles of HBP in vivo and in vitro suggest that more thorough systematic approaches using in vivo systems are needed to determine the role of HBP and the mechanism involved in insulin resistance.
- Ectopic lipid accumulation
- Obesity is an obvious and significant risk factor of T2DM, but numerous studies have shown that ectopic lipid accumulation in peripheral tissues, especially in liver and skeletal muscle, can lead to more severe insulin resistance, even in the absence of visceral adiposity [81]. Consistently, over 70% of obese individuals with T2DM are known to have NAFLD, and NAFLD patients almost universally have T2DM and hepatic insulin resistance [82]. In addition, interventions that reduce intrahepatic triglyceride content through modest weight loss or leptin treatment dramatically reverse hepatic insulin resistance in T2DM, NAFLD, and lipodystrophic patients [83]. Furthermore, intrahepatic and intramyocellular lipid contents are considered to be much better predictors of insulin resistance than visceral adipose tissue volume, which strongly indicates that increased lipid accumulation in liver and skeletal muscle could impair insulin signaling and induce insulin resistance [84,85].
- Numerous animal experiments support the notion that insulin resistance is caused by ectopic lipid accumulation in liver or skeletal muscle. Several studies have shown that lipid accumulation in liver and skeletal muscle caused by short-term HFD feeding or lipid/heparin infusions induce insulin resistance in rats [86]. In addition, overexpression of lipoprotein lipase (LPL) in liver or muscle induced peripheral insulin resistance and the accumulation of lipid in respective tissues [87,88], and skeletal muscle-specific LPL deletion enhanced insulin signaling in HFD challenged muscle [89]. Furthermore, deleting fat transport proteins such as CD36 or FATP-1 increased insulin-mediated glucose uptake in skeletal muscle [90,91], and liver-specific knockdown of FATP2 or FATP5 significantly reduced HFD-induced hepatosteatosis and increased glucose tolerance [92,93].
- Lipodystrophy provides the most well-established clinical evidence that sheds light on the role of ectopic lipid accumulation in the absence of any contribution from visceral adipose to insulin resistance, as this condition is characterized by a substantial reduction in fat cells in adipose tissue leading to hypertriglyceridemia, peripheral ectopic fat deposition, and severe insulin resistance. In lipodystrophy, postprandial fatty acid influx cannot be buffered by adipose tissue, and as a result fatty acids are delivered to other metabolic tissues, including liver and skeletal muscle, which impairs insulin signaling and causes severe insulin resistance [94]. Genetic lipodystrophic mice also exhibit insulin resistance induced by ectopic lipid accumulation in liver and skeletal muscle. Adipocyte-specific basic region-leucine zipper (B-ZIP) transcription factor knockout mice, which are called A-ZIP/F-1 fatless mice due to a lack of white fat tissue, are hyperinsulinemic and hyperglycemic, due to severe defects in IRS-1 and -2 associated PI3K activity in muscle and liver [95,96]. In addition, insulin functions in A-ZIP/F-1 fatless mice were dramatically restored by the transplantation of parametrial fat from littermates, suggesting that the distribution of triglyceride to skeletal muscle and liver might be a critical factor of insulin resistance [95]. The overexpression of preadipocyte factor-1 (Pref-1), a secreted protein that inhibits adipocyte differentiation, also induced the characteristics of lipodystrophic models, that is, reduced adipose tissue mass, dyslipidemia, and insulin resistance [97,98]. In addition, the inhibition of de novo sphingolipid biosynthesis by adipocyte-specific knockout of serine palmitoyltransferase 2 (Sptlc2), which catalyzes the first step of de novo sphingolipid synthesis, exhibited impaired adipose tissue development and a lipodystrophic phenotype, which progressed to systemic insulin resistance [99]. The ability to form unilocular lipid droplets in white adipocytes is required to maintain the ability of white adipocytes to store lipids. Knock out mice of fat-specific protein 27 (Fsp27) showed multilocular lipid droplets in adipocytes and increased lipolysis, resulting in hepatic steatosis and insulin resistance [100]. These studies demonstrate that excess lipid accumulation in liver and muscle can induce insulin resistance.
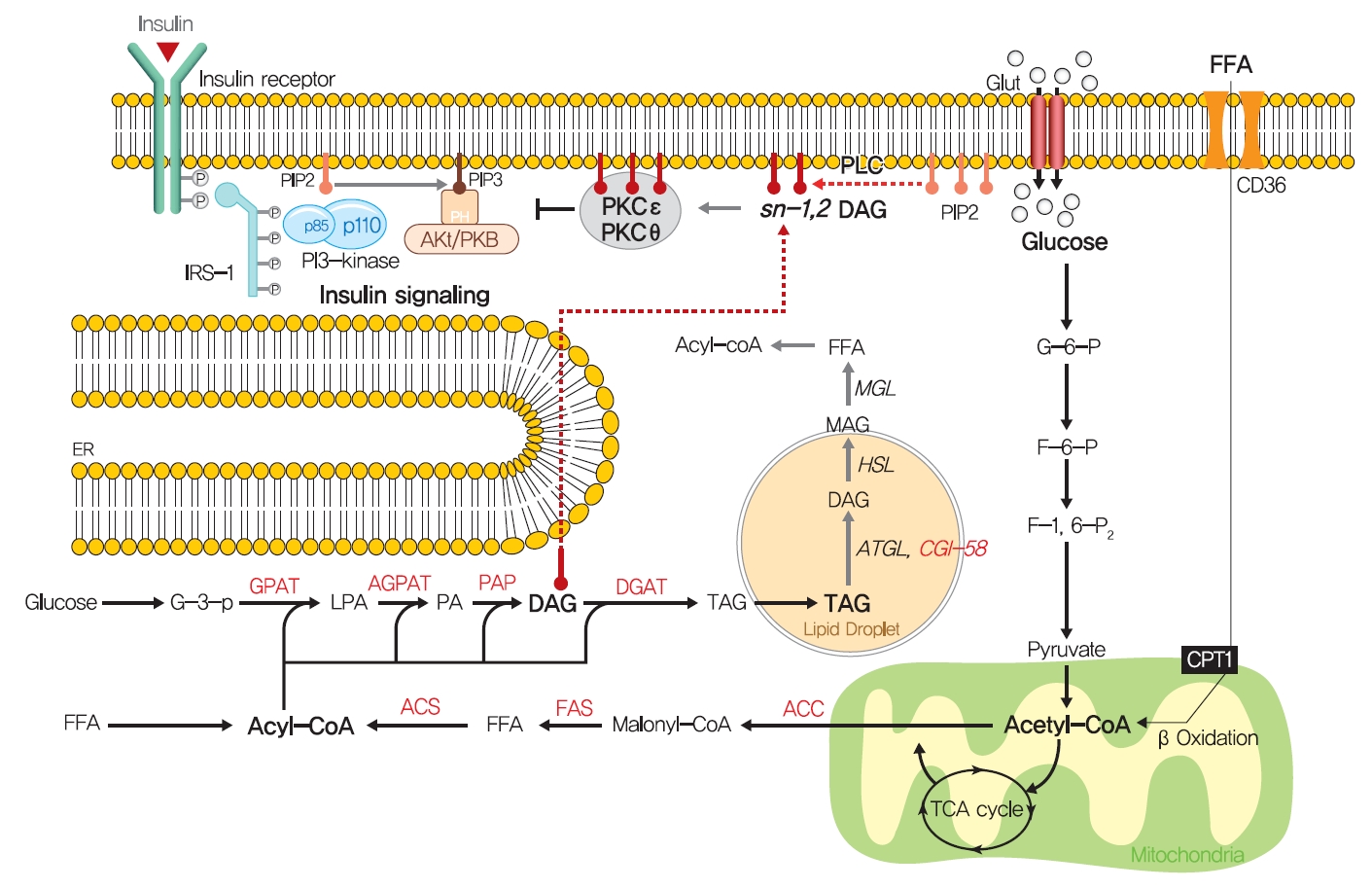
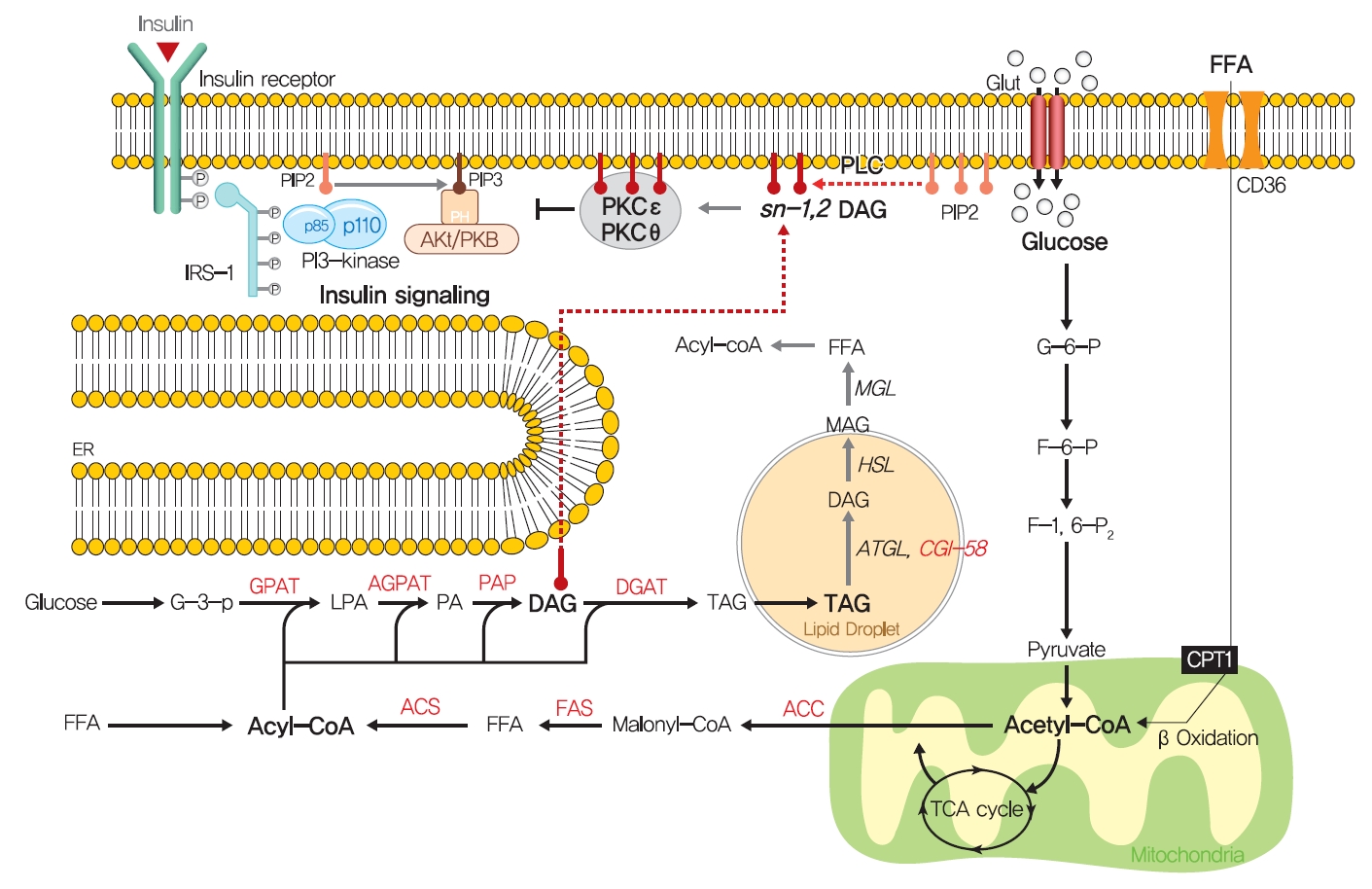
- How excess lipid accumulation interferes with insulin signaling in liver and muscle has long been investigated, especially with respect to surface insulin receptor downregulation and impaired insulin signal transduction in typical obesity-associated insulin resistance [101]. The most plausible hypothesis of the mechanism whereby ectopic lipid accumulation induces insulin resistance is that several lipid metabolites, including diacylglycerol (DAG), lysophosphatidic acid (LPA), ceramides, and acylcarnitines, are involved in the pathogenesis of insulin resistance in liver and skeletal muscle.
- Fatty acids are rapidly esterified in cells to fatty acyl-CoA, which is transferred to a glycerol backbone to form LPA, DAGs, and triacylglycerols (TAGs) through lipogenesis (Fig. 4). These lipid intermediates, especially DAG, might function as second messengers in key signaling pathways implicated in the pathogenesis of insulin resistance [102]. High-fat-induced and genetically obese rodents exhibited hepatic insulin resistance and elevated hepatic DAG contents [103,104], and hepatocytes treated with phorbol myristate acetate (PMA, a DAG analog) exhibited impaired IRTK tyrosine kinase activity and insulin-stimulated GYS activity [105].
- The DAG hypothesis of lipid-induced insulin resistance is that interference of insulin signaling by activated protein kinase C (PKC) results from the accumulation of DAG within insulin-sensitive tissues (Fig. 4). There are three groups of the PKC family (conventional, novel, and atypical), and novel PKC (nPKC), which has much greater affinity for DAG, is known to mediate the role of DAG in insulin resistance [106,107]. In liver, increased hepatic DAG levels induced the translocation of PKCε (the primary nPKC isoform in liver) to the plasma membrane and inhibited IRTK tyrosine kinase activity by phosphorylating it at Thr1160, which reduced insulin-stimulated phosphorylations (activations) of IRS2, PI3K, and Akt2 [108,109]. In the same vein, hepatic DAG contents and PKCε translocation to the plasma membrane were shown to be more potent predictors of hepatic insulin resistance than other factors such as ceramide, endoplasmic reticulum (ER) stress marker, and inflammatory cytokine levels in humans [106,110,111]. In a manner similar to that observed in liver, the accumulation of intramyocellular DAG impairs insulin signaling and muscle glucose uptake by activating PKCθ (muscle-type nPKC) [112,113], which elicits the phosphorylation of IRS-1 at Ser1101 and blocks the insulin-stimulated phosphorylation of IRS-1 [114,115]. Large-scale studies have also corroborated the DAG-PKC induced hypothesis of insulin resistance. High PKC levels have been reported in the membrane fractions of soleus muscles of diabetic rats and in the livers of obese rats [106,116]. Importantly, PKCε translocation to the plasma membrane was observed in rats with isolated hepatic steatosis and hepatic insulin resistance induced by HFD feeding [86,117]. Moreover, knockdown of PKCε using an antisense oligonucleotide (ASO) protected rats from fat-induced hepatic insulin resistance [109], and PKCε knockout mice (Prkce−/−) mice showed improved glucose tolerance than wild type controls after 7 days of high-fat feeding [118]. Apolipoprotein CIII (ApoC3) overexpression promoted diet-induced hepatic steatosis and hepatic insulin resistance, and resulted in higher DAG and membrane-localized PKCε [119]. Like PKCε, PKCθ modulated insulin signaling in myoblasts, and PKC-θ knockout prevented fat-induced defects in insulin signaling and glucose transport in mouse skeletal muscle [120]. Furthermore, PKCθ overexpression led to insulin resistance characterized by reduced insulin responsiveness in C2C12 myoblasts [121].
- Notably, not all DAGs can stimulate PKC translocation to the plasma membrane and induce T2DM by inhibiting insulin signaling. The subcellular localizations of DAGs and DAG stereoisomer types were considered critical for activating PKC and inducing insulin resistance. Of the three DAG stereoisomers, sn-1,2-DAG, the product of the action of phospholipase C on PIP2, was reported to be the only stereoisomer that activate PKC [122]. Furthermore, the accumulation of DAG in lipid droplets, but not in plasma membrane, did not inhibit insulin signaling or induce hepatic insulin resistance, as shown in mice treated with adipose triglyceride lipase cofactor (comparative gene identification-58 [CGI-58]) ASO. CGI-58 ASO treated mice had increased hepatic DAG contents, but preserved hepatic insulin sensitivity [123]. Consequently, sn-1,2-DAG distribution in the plasma membrane disrupted insulin signaling and contributed to insulin resistance through PKC activation [123,124].
- Ceramide is a sphingolipid and an essential bioactive lipid produced from an intracellular fatty acid and sphingosine and is believed to mediate lipid-induced insulin resistance [125]. Ceramide plays a crucial role in cell membrane stabilization and also regulates the distributions of signaling molecules. In obese humans, hepatic ceramides are known to be associated with homeostatic model assessment for insulin resistance (HOMA-IR) scores [126], and obese rats with insulin resistance have consistently been reported to exhibit elevated hepatic and muscle ceramide contents [103]. Inhibition of ceramide synthesis using myriocin, an inhibitor of serine palmitoyltransferase, prevented insulin resistance and attenuated ceramide contents in fat-fed mice [127,128]. In addition, mice heterozygous for dihydroceramide desaturase 1 (Des1), a gatekeeper of ceramide synthesis, had low fasting HOMA-IR scores and total ceramide levels in liver [127]. Also, liver-specific knock out of ceramide synthase 6 (CerS6) decreased hepatic ceramide levels (especially C16:0 species), protected against HFD-induced obesity, and improved glucose tolerance [129]. In addition, liver-specific overexpression of ceramidase protected HFD-fed mice against hepatic steatosis, enhanced hepatic insulin signaling, and reduced the levels of some ceramide species, including 16:0, 18:0, and 20:0 ceramides [130]. Furthermore, adipose- or liver-specific adiponectin receptor (AdipoR) overexpression reduced total ceramide contents and prevented HFD-induced insulin resistance [131]. The results of these studies suggest that ceramide levels influence lipid-induced insulin resistance.
- The underlying molecular mechanism whereby ceramide induces insulin resistance has not been clearly demonstrated. However, some candidate mechanisms have been proposed, namely, the impairment of AKT translocation through the activation of atypical PKCζ, and the activation of PP2A [132,133]. Overexpression of ceramidase in liver and adipose tissue reduced PKCζ activity and prevented ceramide-induced insulin resistance and hepatic steatosis [130]. However, the ceramide analog, C2-ceramide, activated PP2A but not PKCζ and inactivated Akt activity in brown adipocytes [134], which suggested PP2A might mediate the role of ceramide in insulin resistance. However, LB1 (a PP2A inhibitor) increased hepatic insulin resistance as evidenced by elevated fasting plasma glucose and decreased the glucose infusion rate during hyperinsulinemic-euglycemic clamp testing in rats, despite strengthening hepatic insulin signaling, which indicated PP2A activation is not a major ceramide pathway to insulin resistance [135]. Furthermore, fibroblast growth factor 21 (FGF21), adiponectin, and NOD-like receptor pyrin domain-containing protein-3 (NLRP-3) inflammasome have been proposed to mediate or be associated with the function of ceramide [136-138]. Taken together, although growing evidence suggests ceramide can induce insulin resistance, further studies are needed to elucidate the molecular mechanism whereby ceramides impair insulin signaling or glucose transport translocation.
- Other mechanisms: ER stress and inflammation
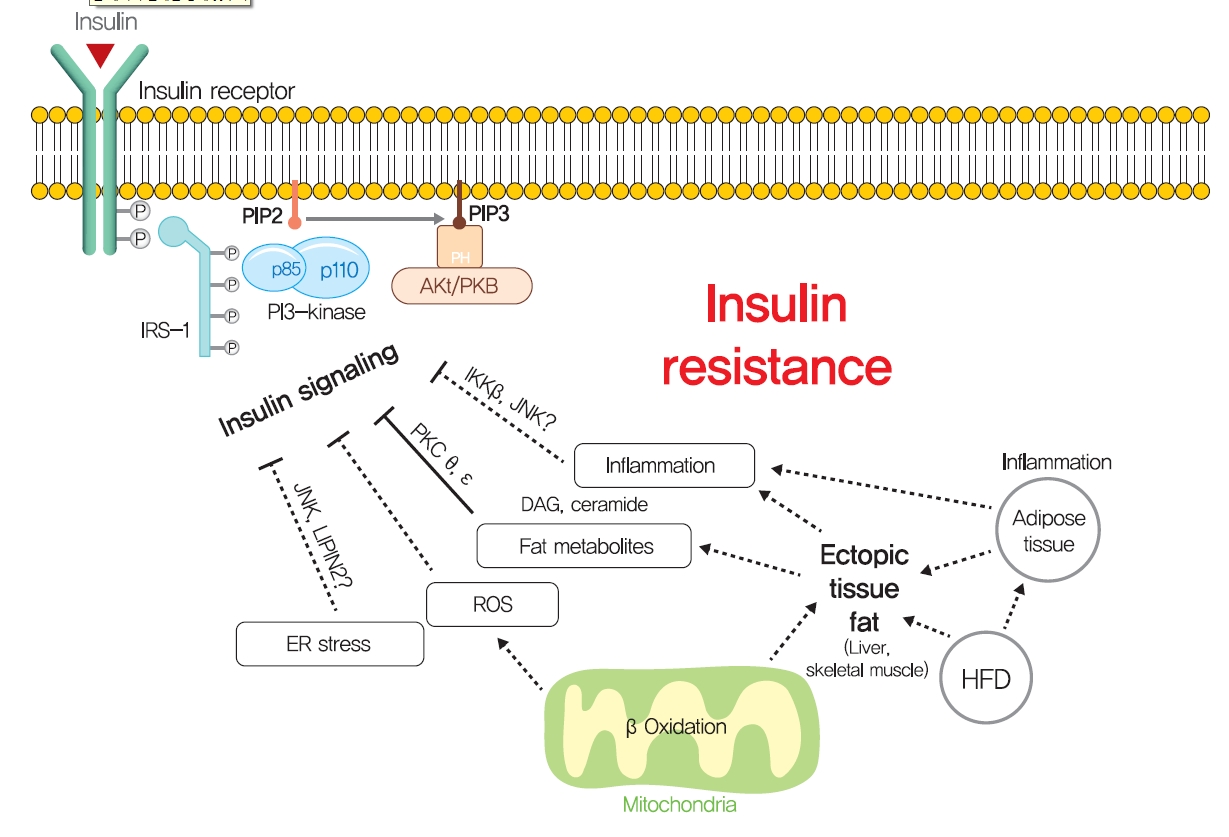
- Although ectopic lipid accumulation in muscle and liver plausibly explains the development of insulin resistance in obese or dyslipidemic subjects, several other hypotheses also have been proposed to explain the mechanism responsible for obesity-induced insulin resistance (Fig. 5). In particular, ER stress has been proposed to underlie insulin resistance in liver and pancreatic β-cells. ER stress is known to be enhanced by obesity [139]. When exposed to an over-nourished condition, the liver should produce an excess of enzymes to process nutrients and enhance unfolded protein response (UPR) by accumulating unfolded proteins in ER. During this process, the recruitment of glucose-regulated protein 78 (GPR78, also known as BiP) results in the activations of inositol requiring enzyme-1 (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6), which inhibit protein translation, promote ER chaperones, and thus, reduce unfolded protein levels [140]. Experimentally, ER stress induced by tunicamycin suppresses insulin signaling via the serine phosphorylation of IRS-1 by c-Jun N-terminal kinase (JNK). In addition, the oral administration of chemical chaperones, such as 4-phenyl butyric acid or taurine-conjugated ursodeoxycholic acid, that alleviate ER stress, improved insulin signaling and restored insulin sensitivity in ob/ob mice [141]. As occurs in liver, increased demand for insulin secretion induces ER stress in pancreatic β-cells in chronic hyperglycemic diabetic humans and mice, and this contributes to the development of T2DM [142,143]. Pancreatic β-cell-specific knockout of X-box-binding protein-1 (XBP-1), a key transcription factor in ER stress, resulted in hyperglycemia and diet-induced insulin resistance resulting from β-cell dysfunction in mice [139,144]. Thus, it appears that, it is premature to conclude that ER stress directly induces insulin resistance, although some aspects of ER stress seem to regulate glucose and lipid metabolism, such as lipogenesis, lipid droplet formation, and lipid storage.
- Systemic chronic inflammatory response characterized by altered cytokine production and the activations of inflammatory signaling pathways is being actively investigated to determine its role in obesity-related insulin resistance [145]. Inflammation-related cytokines, such as tumor necrosis factor-α (TNFα), have been reported to be excreted by macrophages recruited by adipocytes in obese and diabetic humans and animals [146,147]. When macrophages are recruited by adipose tissue due to the overexpression of monocyte chemoattractant protein 1 (MCP1, a chemokine ligand also known as CCL2), hepatic insulin resistance is observed with increased TNFα expression in adipose tissue, but without any change in body weight or adiposity [148]. In addition, knockout of MCP1 protected against HFD-induced insulin resistance [148]. Consistently, knockout of CCR2 (the receptor of MCP-1) and treatment with INCB3344 (a CCR2 antagonist), increased insulin sensitivity and reduced macrophage recruitment by adipose tissue [149]. The role of TNFα in insulin resistance is considered to be mediated by the JNK1, which phosphorylates serine 307 of IRS1 [150]. TNFα also activates inhibitor of nuclear factor κ-B kinase (IKK), and liver- or myeloid cell-specific deletion of IKK protected against hepatic insulin resistance, improved hepatic insulin signaling, and reduced inflammatory cytokine expression [151], which suggested IKK might mediate the role of TNFα in insulin resistance. These studies support the role of inflammation, at least the involvements of TNFα and MCP1, in insulin resistance.
- However, liver-specific knockout of NF-κB essential modulator (NEMO) led to IKK activation, lowered fasting plasma glucose and fasting insulin, improved glucose tolerance, and increased inflammation in liver [152]. In addition, the overexpression of the NF-κB p65 subunit somewhat protected against diet-induced insulin resistance and improved hepatic and peripheral insulin sensitivities [153]. Furthermore, plasma levels of inflammatory cytokines, such as TNFα and interleukin-6, were unchanged in insulin resistant offspring of T2DM and were similar in insulin resistant elderly subjects compared with insulin-sensitive controls [154,155]. These studies demonstrate that chronic inflammation is not a primary causative factor of insulin resistance and that it is insufficient to disrupt systemic glucose metabolism. Thus, it seems that chronic inflammation indirectly exacerbates insulin resistance, and should not be considered a primary strategic target for insulin resistance and T2DM. Rigorously controlled additional studies are required to better determine the role of chronic inflammation in insulin resistance and glucose metabolism.
MECHANISMS OF INSULIN RESISTANCE
DAG
Ceramide
ER stress
Inflammation
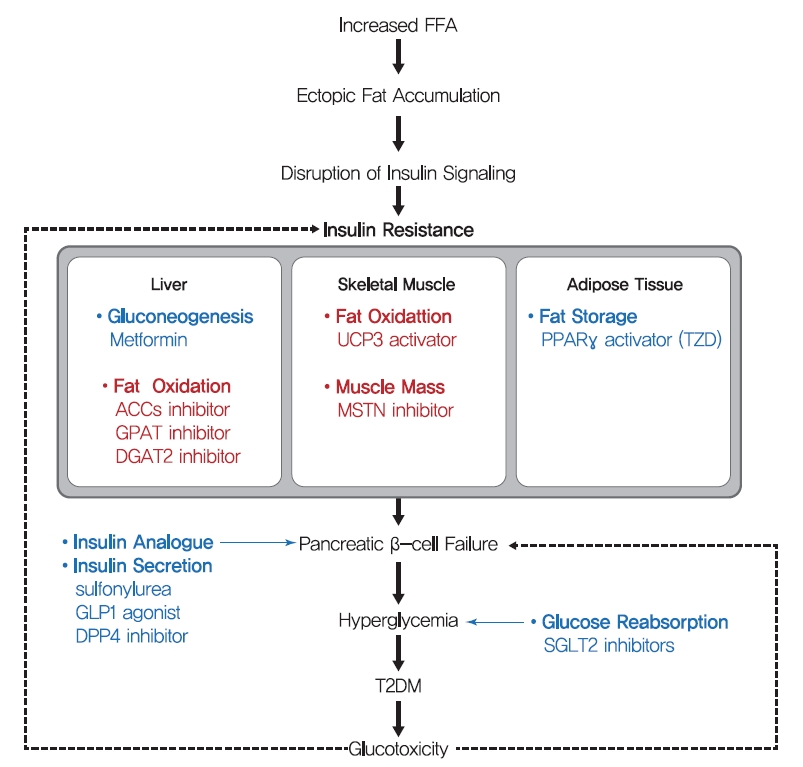
- Pharmaceutical drugs for T2DM treatment
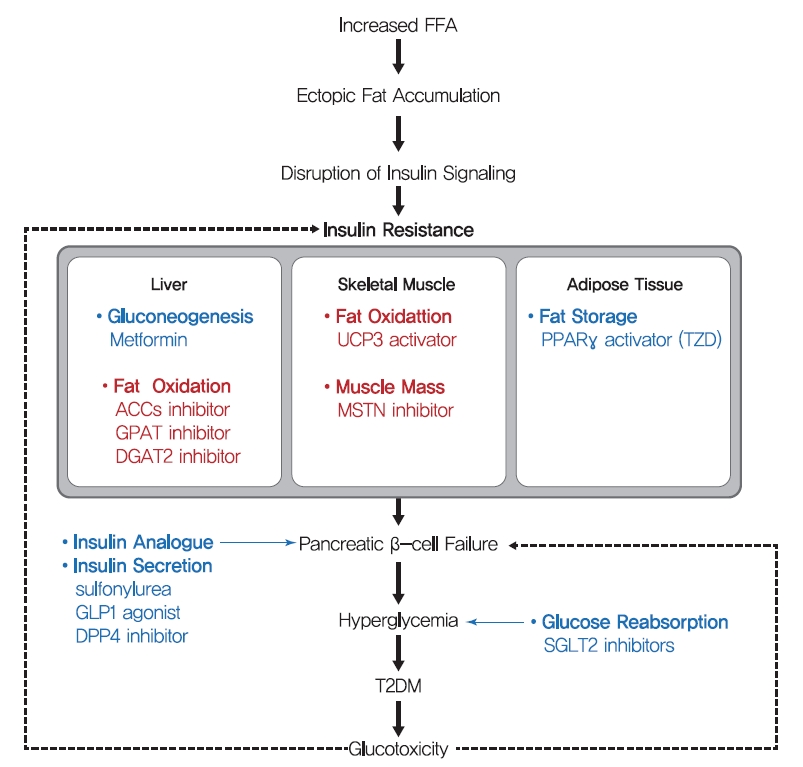
- One hundred years ago, diabetes was considered a dreadful disease accompanied by continued weight loss, emaciation, ketoacidosis, and ultimately coma and death. After the use of insulin as an antidiabetic, diabetes-related mortality has dramatically reduced. However, sedentary lifestyles and the increased prevalence of obesity have increased the incidence of T2DM [156]. Although lifestyle modifications and weight loss are recommended to ameliorate T2DM, their effects are limited with low feasibility. Drugs currently prescribed for T2DM either stimulate insulin secretion or increase insulin sensitivity (Fig. 6). Since the currently available pharmaceutical drugs for T2DM treatment has been well described in many reviews, we briefly summarized the representative T2DM drugs in this review.
- β-Cell dysfunction is a critical feature of the shift from prediabetes to T2DM [157], and many T2DM drugs target the ability of β-cells to secret insulin. Sulfonylureas stimulate β-cells to release insulin independently of glucose by blocking sulphonylurea receptor-1 (SUR-1), which is a regulatory subunit of ATP-sensitive K+ channel in β-cells [158]. In addition, glucagon-like peptide-1 (GLP-1), a hormone produced by L-cells in small intestine in response to food intake, stimulates the production and secretion of insulin by pancreatic β-cells and inhibits glucagon secretion by pancreatic α-cells. Since GLP-1 has poor stability, GLP-1 agonists with longer half-lives and dipeptidyl peptide-4 (DPP-4) inhibitors are used to treat T2DM. Although these drugs stimulate insulin secretion by β-cells to improve hyperglycemia and reduce the risks posed by glucotoxicity, they do not prevent disease progression.
- Thiazolidinediones (TZDs) (such as pioglitazone and rosiglitazone) enhance insulin sensitivity in skeletal muscle, liver, and adipose tissue and promote fat redistribution from liver and skeletal muscle to adipocytes [159]. The effect of TZDs on insulin sensitivity is mediated by the activation of PPARγ, a nuclear receptor that regulates the transcriptions of several genes involved in glucose and lipid metabolism and energy balance and increases fat oxidation, adipocyte proliferation, lipogenesis, fat redistribution, and reduces plasma free fatty acid and pro-inflammatory cytokine levels [159]. Metformin, the most commonly prescribed drug for diabetes, may also increase insulin sensitivity in peripheral tissues during fasting by reducing HGP, which is considered to be mediated by the promotion of mitochondrial activity or the inhibition of glucagon signaling through AMPK activation [160,161].
- In addition to drugs that increase insulin secretion and sensitivity, some T2DM drugs use other strategies. Alpha-glucosidase inhibitors (AGIs) inhibit gastrointestinal hydrolyzing enzymes, which delay the absorption of carbohydrates in small intestine and decrease postprandial blood glucose and insulin levels [162]. On the other hand, sodium-glucose cotransporter 2 (SGLT2) inhibitors lower blood glucose levels by blocking renal glucose reabsorption, which reduces glucotoxicity, improves β-cell function, and increases insulin sensitivity [163].
- As indicated above, many drugs are prescribed to treat diabetes, and the enormous medical expenses associated with the development of diabetes treatments continue to burden pharmaceutical companies. Nevertheless, over recent years the prevalence and mortality of diabetes have increased rapidly. Considering the main causes of the development of T2DM, impaired insulin secretion and insulin resistance, the apparent inefficiency of current pharmaceutical strategies may be due to the undue focus place on alleviating symptoms, such as hyperglycemia by stimulating insulin secretion. Moreover, only two drugs targeting insulin resistance are available, while there are many drugs targeting insulin secretion [164]. Evidence shows insulin resistance is preceding and central to the development of T2DM and is mainly driven by ectopic fat accumulation in tissues. Moreover, insulin resistance is a common pathogenic component of the development of metabolic syndrome, which is strongly associated with obesity. Accordingly, we suggest different therapeutic strategies be adopted that target insulin resistance, and we believe this approach provides a more effective means of preventing and treating diabetes and metabolic syndrome.
- Therapeutic strategy for insulin resistance
- As mentioned above, the accumulation of lipid in hepatocytes induces hepatic insulin resistance via the activation of PKCε by hepatic DAG. Since liver function is central to glucose and lipid metabolism control, hepatic insulin resistance leads to systemic insulin resistance. Hence, the suppression of lipid synthesis through de novo lipogenesis and re-esterification are thought to constitute an effective means of reducing lipid accumulation in liver. This opinion is well supported by the results of animal studies. ACC1 and ACC2 are critical regulators of de novo lipogenesis and lipid oxidation, respectively. Inhibitions of ACC1 and ACC2 using ASO reduced liver DAG contents and PKCε translocation to the plasma membrane and protected mice from lipid-induced hepatic insulin resistance [165]. In addition, the adenovirus-induced overexpression of mitochondrial glycerol-3-phosphate acyltransferase (mGPAT, a lipogenesis rate-limiting enzyme) led to insulin resistance and increased DAG levels in hepatocytes [166]. In the same vein, mGPAT knockout mice had reduced hepatic DAG levels and were protected from HFD-induced hepatic insulin resistance [167], and shRNA knockdown of phosphatidic acid phosphatases (PAPs, also called Lipins) similarly reduced hepatic DAG levels and improved insulin sensitivity, while the overexpression of Lipin-2 using adenovirus perturbed glucose tolerance and insulin sensitivity [168,169]. Also, suppression of diacylglycerol acyltransferase 2 (DGAT2, an enzyme that catalyzes the final step of triglyceride synthesis from diacylglycerol) decreased hepatic DAG contents, reversed diet-induced hepatic steatosis and insulin resistance, and reduced PKCε levels in plasma membranes [170]. These studies strongly support the suggestion that therapies which reduce hepatic DAG and acetyl-CoA contents by suppressing hepatic de novo lipogenesis might effectively treat hepatic insulin resistance in T2DM. In this strategy, agents targeting ACC1, ACC2, and DGAT2 are in phase II clinical trials for the treatment of T2DM and nonalcoholic steatohepatitis (Table 1) [171].
- While the liver is qualitatively critical for glucose metabolism, skeletal muscle is quantitatively essential since skeletal muscle accounts for 35%–45% of body weight and up to 70%–80% of total glucose disposal after insulin stimulation. Therefore, increasing fat oxidation and/or skeletal muscle mass are considered potential strategies for reducing intramyocellular lipid accumulation, and eventually, improving insulin sensitivity. Skeletal muscle-specific overexpression of uncoupling protein 3 (UCP-3, an inner mitochondrial membrane transporter that dissipates mitochondrial proton gradients) protected against HFD-induced insulin resistance in skeletal muscle, enhanced energy expenditure, and reduced PKCθ activity and membrane-associated DAG levels [172]. The beneficial effect of fat oxidation in muscle on insulin resistance was also observed in ACC-2 (a key regulator of mitochondrial fat oxidation) knocked down mice [173]. In addition, skeletal muscle-specific deletion of autophagy-related gene-7 (ATG-7) protected mice from diet-induced obesity and insulin resistance and was accompanied by increased fat oxidation and adipocyte browning [174]. Also, skeletal muscle-specific overexpression of PPARδ improved glucose tolerance and increased number of slow-twitch muscle fibers (a mitochondria rich fiber type) in which mitochondrial oxidative metabolism contributes to energy production [175]. Furthermore, increased number of fast-twitch muscle fibers induced by myostatin (MSTN, also known as Gdf-8) ablation or the skeletal muscle-specific expression of a constitutively active form of Akt1 prevented dieti-nduced obesity, enhanced peripheral insulin sensitivity, and dramatically improved diet-induced insulin resistance [176], which were probably caused by increased glucose utilization and fat oxidation due to increased muscle mass. Despite the beneficial effects of increased muscle mass and quality improve glucose metabolism, few diabetic drugs under development target muscle. Monoclonal antibodies for MSTN are in phase I clinical trials for the treatment of metabolic disorders and cardiovascular disease (Table 1). As regards the developmental efforts to increase muscle quality, bezafibrate, which has been used to treat liver cirrhosis and targets PPARs, is the subject of a phase II clinical trial on T2DM, and chiglitazar sodium, a PPAR pan-agonist, recently approved in China for the treatment of T2DM (Table 1) [171].
THERAPEUTIC STRATEGIES FOR INSULIN RESISTANCE
Inhibiting fat synthesis in liver
Stimulation of fat oxidation and muscle mass
- According to the World Health Organization, the number of people with diabetes rose almost four-fold between 1980 and 2019 from 108 to 463 million, and this increase was accompanied by a steady increase in the prevalence of obesity. In addition, premature mortality attributed to diabetes increased by 5% between 2000 and 2016. In the general populations of most countries, the prevalence of hyperglycemia lies between 7% and 14% [156]. Furthermore, the global prevalence of diabetes has been forecast to increase to 700 million by 2045, and the rapidly increasing prevalence of metabolic disease is now viewed as a global health emergency [164].
- As a primary and common cause of metabolic disease, insulin resistance should be considered a therapeutic target for metabolic diseases, including diabetes. However, no generally accepted theory explains the mechanism responsible for insulin resistance. Nevertheless, growing evidence demonstrates that ectopic lipid accumulation is more strongly related to diabetic physiology than other variables, such as ER stress and the plasma concentrations of inflammatory cytokine [110,111,126]. Numerous animal and epidemiological studies have shown that DAG accumulation in plasm membrane fractions caused by ectopic fat accumulation or reduced fat oxidation is a critical factor of insulin resistance development via the activation of nPKC in liver tissues and skeletal muscle. Based on this pathophysiological mechanism of insulin resistance, the suppression of lipid synthesis in liver and the stimulation of fat oxidation in skeletal muscle provide means of reducing ectopic lipid accumulation and potentially, improving insulin sensitivity, and eventually, preventing or delaying the onset of T2DM.
- Like other metabolic diseases, diabetes is a multifaceted disease. Although ectopic lipid accumulation in peripheral tissue appears to be the primary cause of insulin resistance, increased ER stress, inflammation, and reactive oxygen species levels, dysregulations of adipokines contribute to the pathogenesis of insulin resistance in liver and skeletal muscle. Therefore, we suggest that treatments for insulin resistance should be based on multidisciplinary strategies that target physiological and metabolic impairments.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No.2014M3A9D5A01073886) and by the Gachon University research fund of 2018 (GCU-2018-0683).
NOTES
-
Acknowledgements
- None


Data from database Cortellis Drug Discovery Intelligence (Clarivate Analytics) [171].
ACC, acetyl-CoA carboxylase; DGAT2, diacylglycerol acyltransferase 2; PPAR, peroxisome proliferator-activated receptor; MSTN, myostatin.
- 1. Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001;414:782-7.ArticlePubMedPDF
- 2. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 2003;46:3-19.ArticlePubMedPDF
- 3. Rizza RA. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes 2010;59:2697-707.ArticlePubMedPMCPDF
- 4. Moore MC, Cherrington AD, Cline G, Pagliassotti MJ, Jones EM, Neal DW, et al. Sources of carbon for hepatic glycogen synthesis in the conscious dog. J Clin Invest 1991;88:578-87.ArticlePubMedPMC
- 5. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev 2018;98:2133-223.ArticlePubMedPMC
- 6. Asplin CM, Paquette TL, Palmer JP. In vivo inhibition of glucagon secretion by paracrine beta cell activity in man. J Clin Invest 1981;68:314-8.ArticlePubMedPMC
- 7. Greenbaum CJ, Havel PJ, Taborsky GJ Jr, Klaff LJ. Intra-islet insulin permits glucose to directly suppress pancreatic A cell function. J Clin Invest 1991;88:767-73.ArticlePubMedPMC
- 8. Youngren JF. Regulation of insulin receptor function. Cell Mol Life Sci 2007;64:873-91.ArticlePubMedPDF
- 9. Khalid M, Alkaabi J, Khan MAB, Adem A. Insulin signal transduction perturbations in insulin resistance. Int J Mol Sci 2021;22:8590.ArticlePubMedPMC
- 10. Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 2012;13:383-96.ArticlePubMedPDF
- 11. Chiu TT, Jensen TE, Sylow L, Richter EA, Klip A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal 2011;23:1546-54.ArticlePubMed
- 12. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995;378:785-9.ArticlePubMedPDF
- 13. Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes 2000;49:1967-77.ArticlePubMedPDF
- 14. Agius L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med 2015;46:34-45.ArticlePubMed
- 15. Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab 2008;8:65-76.ArticlePubMedPMC
- 16. Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors: regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 2011;1813:1938-45.ArticlePubMed
- 17. Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015;160:745-58.ArticlePubMedPMC
- 18. Ros S, Garcia-Rocha M, Dominguez J, Ferrer JC, Guinovart JJ. Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. J Biol Chem 2009;284:6370-8.ArticlePubMed
- 19. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125-31.ArticlePubMedPMC
- 20. Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab 2010;21:268-76.ArticlePubMed
- 21. Rebrin K, Steil GM, Mittelman SD, Bergman RN. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest 1996;98:741-9.ArticlePubMedPMC
- 22. Choi YH, Park S, Hockman S, Zmuda-Trzebiatowska E, Svennelid F, Haluzik M, et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J Clin Invest 2006;116:3240-51.ArticlePubMedPMC
- 23. Begum N. Stimulation of protein phosphatase-1 activity by insulin in rat adipocytes. Evaluation of the role of mitogen-activated protein kinase pathway. J Biol Chem 1995;270:709-14.PubMed
- 24. Resjo S, Goransson O, Harndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal 2002;14:231-8.ArticlePubMed
- 25. Virtanen KA, Lonnroth P, Parkkola R, Peltoniemi P, Asola M, Viljanen T, et al. Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J Clin Endocrinol Metab 2002;87:3902-10.ArticlePubMed
- 26. Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep 2001;2:282-6.ArticlePubMedPMCPDF
- 27. Rieusset J, Andreelli F, Auboeuf D, Roques M, Vallier P, Riou JP, et al. Insulin acutely regulates the expression of the peroxisome proliferator-activated receptor-gamma in human adipocytes. Diabetes 1999;48:699-705.ArticlePubMedPDF
- 28. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009;32 Suppl 2:S157-63.ArticlePubMedPDF
- 29. Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest 2000;105:1791-7.ArticlePubMedPMC
- 30. Kim JK, Zisman A, Fillmore JJ, Peroni OD, Kotani K, Perret P, et al. Glucose toxicity and the development of diabetes in mice with muscle-specific inactivation of GLUT4. J Clin Invest 2001;108:153-60.ArticlePubMedPMC
- 31. Wojtaszewski JF, Higaki Y, Hirshman MF, Michael MD, Dufresne SD, Kahn CR, et al. Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J Clin Invest 1999;104:1257-64.ArticlePubMedPMC
- 32. O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A 2011;108:16092-7.PubMedPMC
- 33. Le Marchand-Brustel Y, Gremeaux T, Ballotti R, Van Obberghen E. Insulin receptor tyrosine kinase is defective in skeletal muscle of insulin-resistant obese mice. Nature 1985;315:676-9.ArticlePubMedPDF
- 34. Frojdo S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochim Biophys Acta 2009;1792:83-92.ArticlePubMed
- 35. Lewis GF, Carpentier AC, Pereira S, Hahn M, Giacca A. Direct and indirect control of hepatic glucose production by insulin. Cell Metab 2021;33:709-20.ArticlePubMed
- 36. Krssak M, Brehm A, Bernroider E, Anderwald C, Nowotny P, Dalla Man C, et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 2004;53:3048-56.ArticlePubMedPDF
- 37. Basu R, Chandramouli V, Dicke B, Landau B, Rizza R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes 2005;54:1942-8.ArticlePubMedPDF
- 38. Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest 1992;90:1323-7.ArticlePubMedPMC
- 39. Wu X, Chen K, Williams KJ. The role of pathway-selective insulin resistance and responsiveness in diabetic dyslipoproteinemia. Curr Opin Lipidol 2012;23:334-44.ArticlePubMed
- 40. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 2010;107:3441-6.ArticlePubMedPMC
- 41. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 2006;11:859-71.PubMed
- 42. Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006;127:125-37.ArticlePubMed
- 43. Kim K, Qiang L, Hayden MS, Sparling DP, Purcell NH, Pajvani UB. mTORC1-independent raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat Commun 2016;7:10255.ArticlePubMedPMCPDF
- 44. Kim K, Ryu D, Dongiovanni P, Ozcan L, Nayak S, Ueberheide B, et al. Degradation of PHLPP2 by KCTD17, via a glucagon-dependent pathway, promotes hepatic steatosis. Gastroenterology 2017;153:1568-80.ArticlePubMed
- 45. Nagai Y, Yonemitsu S, Erion DM, Iwasaki T, Stark R, Weismann D, et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab 2009;9:252-64.PubMedPMC
- 46. Bindesboll C, Fan Q, Norgaard RC, MacPherson L, Ruan HB, Wu J, et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J Lipid Res 2015;56:771-85.ArticlePubMedPMC
- 47. Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr 2007;27:179-92.ArticlePubMed
- 48. Erion DM, Popov V, Hsiao JJ, Vatner D, Mitchell K, Yonemitsu S, et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology 2013;154:36-44.ArticlePubMedPDF
- 49. Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab 2006;4:107-10.ArticlePubMed
- 50. Hauner H. Obesity and diabetes. Chichester: John Wiley & Sons; 2017.ArticlePDF
- 51. Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes 1997;46:1001-9.ArticlePubMed
- 52. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963;1:785-9.ArticlePubMed
- 53. Jucker BM, Rennings AJ, Cline GW, Shulman GI. 13C and 31P NMR studies on the effects of increased plasma free fatty acids on intramuscular glucose metabolism in the awake rat. J Biol Chem 1997;272:10464-73.ArticlePubMed
- 54. Schwartzman LI, Brown J. Glucose inhibition of fatty acid oxidation by rat diaphragm. Am J Physiol 1960;199:235-7.ArticlePubMed
- 55. Christe ME, Rodgers RL. Cardiac glucose and fatty acid oxidation in the streptozotocin-induced diabetic spontaneously hypertensive rat. Hypertension 1995;25:235-41.ArticlePubMed
- 56. Rothman DL, Shulman RG, Shulman GI. 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. Evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J Clin Invest 1992;89:1069-75.ArticlePubMedPMC
- 57. Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med 1999;341:240-6.ArticlePubMed
- 58. Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med 1990;322:223-8.ArticlePubMed
- 59. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem 1991;266:4706-12.ArticlePubMed
- 60. Hawkins M, Barzilai N, Liu R, Hu M, Chen W, Rossetti L. Role of the glucosamine pathway in fat-induced insulin resistance. J Clin Invest 1997;99:2173-82.ArticlePubMedPMC
- 61. Marshall S, Bacote V, Traxinger RR. Complete inhibition of glucose-induced desensitization of the glucose transport system by inhibitors of mRNA synthesis. Evidence for rapid turnover of glutamine:fructose-6-phosphate amidotransferase. J Biol Chem 1991;266:10155-61.ArticlePubMed
- 62. McClain DA. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J Diabetes Complications 2002;16:72-80.ArticlePubMed
- 63. Rossetti L. Perspective: hexosamines and nutrient sensing. Endocrinology 2000;141:1922-5.ArticlePubMed
- 64. Buse MG, Robinson KA, Gettys TW, McMahon EG, Gulve EA. Increased activity of the hexosamine synthesis pathway in muscles of insulin-resistant ob/ob mice. Am J Physiol 1997;272(6 Pt 1):E1080-8.ArticlePubMed
- 65. Robinson KA, Weinstein ML, Lindenmayer GE, Buse MG. Effects of diabetes and hyperglycemia on the hexosamine synthesis pathway in rat muscle and liver. Diabetes 1995;44:1438-46.ArticlePubMed
- 66. Akimoto Y, Hart GW, Wells L, Vosseller K, Yamamoto K, Munetomo E, et al. Elevation of the post-translational modification of proteins by O-linked N-acetylglucosamine leads to deterioration of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto-Kakizaki rats. Glycobiology 2007;17:127-40.ArticlePubMed
- 67. Akimoto Y, Kawakami H, Yamamoto K, Munetomo E, Hida T, Hirano H. Elevated expression of O-GlcNAc-modified proteins and O-GlcNAc transferase in corneas of diabetic Goto-Kakizaki rats. Invest Ophthalmol Vis Sci 2003;44:3802-9.ArticlePubMed
- 68. Hazel M, Cooksey RC, Jones D, Parker G, Neidigh JL, Witherbee B, et al. Activation of the hexosamine signaling pathway in adipose tissue results in decreased serum adiponectin and skeletal muscle insulin resistance. Endocrinology 2004;145:2118-28.ArticlePubMedPDF
- 69. Murata K, Morino K, Ida S, Ohashi N, Lemecha M, Park SY, et al. Lack of O-GlcNAcylation enhances exercise-dependent glucose utilization potentially through AMP-activated protein kinase activation in skeletal muscle. Biochem Biophys Res Commun 2018;495:2098-104.ArticlePubMed
- 70. Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A 2002;99:5313-8.ArticlePubMedPMC
- 71. Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011;60:1399-413.ArticlePubMedPMCPDF
- 72. Farook VS, Bogardus C, Prochazka M. Analysis of MGEA5 on 10q24.1-q24.3 encoding the beta-O-linked N-acetylglucosaminidase as a candidate gene for type 2 diabetes mellitus in Pima Indians. Mol Genet Metab 2002;77:189-93.PubMed
- 73. Lehman DM, Fu DJ, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes 2005;54:1214-21.PubMed
- 74. Copeland RJ, Bullen JW, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab 2008;295:E17-28.ArticlePubMedPMC
- 75. Ball LE, Berkaw MN, Buse MG. Identification of the major site of O-linked beta-N-acetylglucosamine modification in the C terminus of insulin receptor substrate-1. Mol Cell Proteomics 2006;5:313-23.PubMed
- 76. Kang ES, Han D, Park J, Kwak TK, Oh MA, Lee SA, et al. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp Cell Res 2008;314:2238-48.PubMed
- 77. Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, et al. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem 2008;283:16283-92.ArticlePubMedPMC
- 78. Chen G, Liu P, Thurmond DC, Elmendorf JS. Glucosamineinduced insulin resistance is coupled to O-linked glycosylation of Munc18c. FEBS Lett 2003;534:54-60.ArticlePubMedPDF
- 79. Robinson KA, Ball LE, Buse MG. Reduction of O-GlcNAc protein modification does not prevent insulin resistance in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 2007;292:E884-90.ArticlePubMed
- 80. Macauley MS, Bubb AK, Martinez-Fleites C, Davies GJ, Vocadlo DJ. Elevation of global O-GlcNAc levels in 3T3-L1 adipocytes by selective inhibition of O-GlcNAcase does not induce insulin resistance. J Biol Chem 2008;283:34687-95.ArticlePubMedPMC
- 81. Mann JP, Savage DB. What lipodystrophies teach us about the metabolic syndrome. J Clin Invest 2019;129:4009-21.ArticlePubMedPMC
- 82. Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci U S A 2006;103:18273-7.ArticlePubMedPMC
- 83. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005;54:603-8.ArticlePubMedPDF
- 84. Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A 2009;106:15430-5.ArticlePubMedPMC
- 85. Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 1999;42:113-6.ArticlePubMedPDF
- 86. Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 2004;279:32345-53.ArticlePubMed
- 87. Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U S A 2001;98:7522-7.ArticlePubMedPMC
- 88. Merkel M, Weinstock PH, Chajek-Shaul T, Radner H, Yin B, Breslow JL, et al. Lipoprotein lipase expression exclusively in liver. A mouse model for metabolism in the neonatal period and during cachexia. J Clin Invest 1998;102:893-901.ArticlePubMedPMC
- 89. Wang H, Knaub LA, Jensen DR, Young Jung D, Hong EG, Ko HJ, et al. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes 2009;58:116-24.ArticlePubMedPMCPDF
- 90. Goudriaan JR, Dahlmans VE, Teusink B, Ouwens DM, Febbraio M, Maassen JA, et al. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J Lipid Res 2003;44:2270-7.ArticlePubMed
- 91. Kim JK, Gimeno RE, Higashimori T, Kim HJ, Choi H, Punreddy S, et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest 2004;113:756-63.ArticlePubMedPMC
- 92. Doege H, Grimm D, Falcon A, Tsang B, Storm TA, Xu H, et al. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J Biol Chem 2008;283:22186-92.ArticlePubMedPMC
- 93. Falcon A, Doege H, Fluitt A, Tsang B, Watson N, Kay MA, et al. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab 2010;299:E384-93.ArticlePubMedPMC
- 94. Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002;45:1201-10.ArticlePubMedPDF
- 95. Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 2000;275:8456-60.ArticlePubMed
- 96. Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, et al. Life without white fat: a transgenic mouse. Genes Dev 1998;12:3168-81.ArticlePubMedPMC
- 97. Lee K, Villena JA, Moon YS, Kim KH, Lee S, Kang C, et al. Inhibition of adipogenesis and development of glucose intolerance by soluble preadipocyte factor-1 (Pref-1). J Clin Invest 2003;111:453-61.ArticlePubMedPMC
- 98. Villena JA, Choi CS, Wang Y, Kim S, Hwang YJ, Kim YB, et al. Resistance to high-fat diet-induced obesity but exacerbated insulin resistance in mice overexpressing preadipocyte factor-1 (Pref-1): a new model of partial lipodystrophy. Diabetes 2008;57:3258-66.PubMedPMC
- 99. Lee SY, Lee HY, Song JH, Kim GT, Jeon S, Song YJ, et al. Adipocyte-specific deficiency of de novo sphingolipid biosynthesis leads to lipodystrophy and insulin resistance. Diabetes 2017;66:2596-609.ArticlePubMedPMCPDF
- 100. Zhou L, Park SY, Xu L, Xia X, Ye J, Su L, et al. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27-deficient mice. Nat Commun 2015;6:5949.ArticlePubMedPDF
- 101. Carpentier JL. Insulin receptor internalization: molecular mechanisms and physiopathological implications. Diabetologia 1994;37 Suppl 2:S117-24.ArticlePubMedPDF
- 102. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012;148:852-71.ArticlePubMedPMC
- 103. Turinsky J, O’Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem 1990;265:16880-5.ArticlePubMed
- 104. Turner N, Kowalski GM, Leslie SJ, Risis S, Yang C, Lee-Young RS, et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013;56:1638-48.ArticlePubMedPDF
- 105. Takayama S, White MF, Lauris V, Kahn CR. Phorbol esters modulate insulin receptor phosphorylation and insulin action in cultured hepatoma cells. Proc Natl Acad Sci U S A 1984;81:7797-801.ArticlePubMedPMC
- 106. Considine RV, Nyce MR, Allen LE, Morales LM, Triester S, Serrano J, et al. Protein kinase C is increased in the liver of humans and rats with non-insulin-dependent diabetes mellitus: an alteration not due to hyperglycemia. J Clin Invest 1995;95:2938-44.ArticlePubMedPMC
- 107. Karasik A, Rothenberg PL, Yamada K, White MF, Kahn CR. Increased protein kinase C activity is linked to reduced insulin receptor autophosphorylation in liver of starved rats. J Biol Chem 1990;265:10226-31.ArticlePubMed
- 108. Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest 2016;126:4361-71.ArticlePubMedPMC
- 109. Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, et al. Inhibition of protein kinase C epsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 2007;117:739-45.ArticlePubMedPMC
- 110. Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep 2017;19:1997-2004.ArticlePubMedPMC
- 111. Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012;142:1444-6.ArticlePubMed
- 112. Schmitz-Peiffer C, Browne CL, Oakes ND, Watkinson A, Chisholm DJ, Kraegen EW, et al. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes 1997;46:169-78.ArticlePubMed
- 113. Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 2002;277:50230-6.ArticlePubMed
- 114. Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, et al. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem 2004;279:45304-7.PubMed
- 115. Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A 2014;111:9597-602.ArticlePubMedPMC
- 116. Avignon A, Yamada K, Zhou X, Spencer B, Cardona O, Saba-Siddique S, et al. Chronic activation of protein kinase C in soleus muscles and other tissues of insulin-resistant type II diabetic Goto-Kakizaki (GK), obese/aged, and obese/Zucker rats. A mechanism for inhibiting glycogen synthesis. Diabetes 1996;45:1396-404.ArticlePubMed
- 117. Qu X, Seale JP, Donnelly R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats: effects of feeding. J Endocrinol 1999;162:207-14.ArticlePubMed
- 118. Raddatz K, Turner N, Frangioudakis G, Liao BM, Pedersen DJ, Cantley J, et al. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 2011;54:1447-56.ArticlePubMedPDF
- 119. Lee HY, Birkenfeld AL, Jornayvaz FR, Jurczak MJ, Kanda S, Popov V, et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 2011;54:1650-60.ArticlePubMedPDF
- 120. Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest 2004;114:823-7.ArticlePubMedPMC
- 121. Haasch D, Berg C, Clampit JE, Pederson T, Frost L, Kroeger P, et al. PKC theta is a key player in the development of insulin resistance. Biochem Biophys Res Commun 2006;343:361-8.PubMed
- 122. Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun 1984;122:818-23.ArticlePubMed
- 123. Cantley JL, Yoshimura T, Camporez JP, Zhang D, Jornayvaz FR, Kumashiro N, et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci U S A 2013;110:1869-74.ArticlePubMedPMC
- 124. Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res 2010;51:3306-15.ArticlePubMedPMC
- 125. Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 2012;15:585-94.ArticlePubMed
- 126. Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol 2016;64:1167-75.ArticlePubMed
- 127. Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 2007;5:167-79.ArticlePubMed
- 128. Ussher JR, Koves TR, Cadete VJ, Zhang L, Jaswal JS, Swyrd SJ, et al. Inhibition of de novo ceramide synthesis reverses dietinduced insulin resistance and enhances whole-body oxygen consumption. Diabetes 2010;59:2453-64.ArticlePubMedPMCPDF
- 129. Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 2014;20:678-86.ArticlePubMed
- 130. Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab 2015;22:266-78.ArticlePubMedPMC
- 131. Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab 2017;6:267-75.ArticlePubMedPMC
- 132. Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol 2003;23:7794-808.PubMedPMC
- 133. Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem 2004;279:36608-15.PubMed
- 134. Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001;50:2563-71.PubMed
- 135. Galbo T, Perry RJ, Nishimura E, Samuel VT, Quistorff B, Shulman GI. PP2A inhibition results in hepatic insulin resistance despite Akt2 activation. Aging (Albany NY) 2013;5:770-81.ArticlePubMedPMC
- 136. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab 2013;17:790-7.ArticlePubMedPMC
- 137. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 2011;17:55-63.ArticlePubMedPDF
- 138. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011;17:179-88.ArticlePubMedPMCPDF
- 139. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306:457-61.ArticlePubMed
- 140. Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 2002;366(Pt 2):585-94.ArticlePubMedPMCPDF
- 141. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006;313:1137-40.ArticlePubMedPMC
- 142. Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007;50:752-63.ArticlePubMedPDF
- 143. Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007;50:2486-94.ArticlePubMedPDF
- 144. Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci U S A 2011;108:8885-90.PubMedPMC
- 145. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 2005;115:1111-9.ArticlePubMedPMC
- 146. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87-91.ArticlePubMed
- 147. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796-808.ArticlePubMedPMC
- 148. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006;116:1494-505.ArticlePubMedPMC
- 149. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 2006;116:115-24.ArticlePubMed
- 150. Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 2000;275:9047-54.ArticlePubMed
- 151. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191-8.ArticlePubMedPDF
- 152. Wunderlich FT, Luedde T, Singer S, Schmidt-Supprian M, Baumgartl J, Schirmacher P, et al. Hepatic NF-kappa B essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proc Natl Acad Sci U S A 2008;105:1297-302.PubMedPMC
- 153. Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Jung DY, et al. Uncoupling of inflammation and insulin resistance by NF-kappaB in transgenic mice through elevated energy expenditure. J Biol Chem 2010;285:4637-44.PubMed
- 154. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 2004;350:664-71.ArticlePubMedPMC
- 155. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 2003;300:1140-2.ArticlePubMedPMC
- 156. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016;387:1513-30.PubMedPMC
- 157. Boden G. Pathogenesis of type 2 diabetes. Insulin resistance. Endocrinol Metab Clin North Am 2001;30:801-15.PubMed
- 158. Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002;51 Suppl 3:S368-76.ArticlePubMedPDF
- 159. Hauner H. The mode of action of thiazolidinediones. Diabetes Metab Res Rev 2002;18 Suppl 2:S10-5.ArticlePubMed
- 160. Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat Rev Endocrinol 2019;15:569-89.ArticlePubMedPDF
- 161. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 2017;60:1577-85.ArticlePubMedPMCPDF
- 162. Kumar S, Narwal S, Kumar V, Prakash O. α-Glucosidase inhibitors from plants: a natural approach to treat diabetes. Pharmacogn Rev 2011;5:19-29.ArticlePubMedPMC
- 163. Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev 2011;32:515-31.ArticlePubMedPDF
- 164. Guerra J, Dias M, Brilhante A, Terra MF, Garcia-Arevalo M, Figueira A. Multifactorial basis and therapeutic strategies in metabolism-related diseases. Nutrients 2021;13:2830.ArticlePubMedPMC
- 165. Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest 2006;116:817-24.ArticlePubMedPMC
- 166. Nagle CA, An J, Shiota M, Torres TP, Cline GW, Liu ZX, et al. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J Biol Chem 2007;282:14807-15.ArticlePubMed
- 167. Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, Romanelli AJ, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn3-phosphate acyltransferase 1 knockout mice. Cell Metab 2005;2:55-65.ArticlePubMed
- 168. Ryu D, Seo WY, Yoon YS, Kim YN, Kim SS, Kim HJ, et al. Endoplasmic reticulum stress promotes LIPIN2-dependent hepatic insulin resistance. Diabetes 2011;60:1072-81.ArticlePubMedPMCPDF
- 169. Ryu D, Oh KJ, Jo HY, Hedrick S, Kim YN, Hwang YJ, et al. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab 2009;9:240-51.ArticlePubMed
- 170. Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem 2007;282:22678-88.ArticlePubMed
- 171. Clarivate Analytics: Cortellis Drug Discovery Intelligence. Available from: http://www.cortellis.com/drugdiscovery (cited 2021 Dec 16).
- 172. Choi CS, Fillmore JJ, Kim JK, Liu ZX, Kim S, Collier EF, et al. Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J Clin Invest 2007;117:1995-2003.ArticlePubMedPMC
- 173. Choi CS, Savage DB, Abu-Elheiga L, Liu ZX, Kim S, Kulkarni A, et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A 2007;104:16480-5.ArticlePubMedPMC
- 174. Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 2013;19:83-92.ArticlePubMedPDF
- 175. Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol 2004;2:e294.ArticlePubMedPMC
- 176. Wilkes JJ, Lloyd DJ, Gekakis N. Loss-of-function mutation in myostatin reduces tumor necrosis factor alpha production and protects liver against obesity-induced insulin resistance. Diabetes 2009;58:1133-43.PubMedPMC
REFERENCES
Figure & Data
References
Citations
- Diabetic Encephalopathy: Role of Oxidative and Nitrosative Factors in Type 2 Diabetes
Debashree Mazumdar, Santosh Singh
Indian Journal of Clinical Biochemistry.2024; 39(1): 3. CrossRef - Materials and structure of polysaccharide-based delivery carriers for oral insulin: A review
Xinran Wang, Hongnan Sun, Taihua Mu
Carbohydrate Polymers.2024; 323: 121364. CrossRef - β-Thalassemia and Diabetes Mellitus: Current State and Future
Directions
Jalal Taneera, Eglal Mahgoub, Reem Qannita, Ayah Alalami, Ola Al Shehadat, Mona Youssef, Ayah Dib, Alaa Al Hajji, Amani Al Hajji, Fatheya Al-Khaja, Hany Dewedar, Mawieh Hamad
Hormone and Metabolic Research.2024; 56(04): 272. CrossRef - Mulberry leaf multi-components exert hypoglycemic effects through regulation of the PI-3K/Akt insulin signaling pathway in type 2 diabetic rats
Yue Zhang, Liang Li, Tao Chai, Han Xu, Hong-yan Du, Yan Jiang
Journal of Ethnopharmacology.2024; 319: 117307. CrossRef - Thyroid cancer and insulin resistance
Gabriela Brenta, Fernando Di Fermo
Reviews in Endocrine and Metabolic Disorders.2024; 25(1): 19. CrossRef - Comparative profiling of gut microbiota and metabolome in diet-induced obese and insulin-resistant C57BL/6J mice
Hobby Aggarwal, Jyoti Gautam, Deepika Kumari, Sonu Kumar Gupta, Sneh Bajpai, Kartikey Chaturvedi, Yashwant Kumar, Madhu Dikshit
Biochimica et Biophysica Acta (BBA) - Molecular Cell Research.2024; 1871(2): 119643. CrossRef - CD248 promotes insulin resistance by binding to the insulin receptor and dampening its insulin-induced autophosphorylation
Patricia O. Benedet, Nooshin S. Safikhan, Maria J. Pereira, Bryan M. Lum, José Diego Botezelli, Cheng-Hsiang Kuo, Hua-Lin Wu, Barbara P. Craddock, W. Todd Miller, Jan W. Eriksson, Jessica T.Y. Yue, Edward M. Conway
eBioMedicine.2024; 99: 104906. CrossRef - The Antiobesity Effects and Potential Mechanisms of Theaflavins
Yi Fang, Jun Wang, Yu Cao, Wenrui Liu, Lianxiang Duan, Jing Hu, Jinghua Peng
Journal of Medicinal Food.2024; 27(1): 1. CrossRef - The role of zinc finger proteins in the fate determination of mesenchymal stem cells during osteogenic and adipogenic differentiation
Bolun Li, Shibo Liu, Ze He, En Luo, Hanghang Liu
The International Journal of Biochemistry & Cell Biology.2024; 167: 106507. CrossRef - Gentianella turkestanorum (Gand.) Holub, a Chinese Herbal Medicine that can Alleviate T2DM in Db/db Mice, and its Active Mechanism of Action
Ying Wei, Jiaxin Sun, Liya Su, Tunhai Xu
Pharmacognosy Magazine.2024; 20(2): 646. CrossRef - Hyperglycemia affects axial signs in patients with Parkinson’s disease through mechanisms of insulin resistance or non-insulin resistance
Ruidan Wang, Zhaohui Jin, Qiaoxia Zhen, Lin Qi, Cui Liu, Ping Wang, Yonghong Liu, Jinping Fang, Yanjun Liu, Yuan Su, Yixuan Wang, Detao Meng, Hongjiao Yan, Yi Zhen, Zhenzhen Li, Boyan Fang
Neurological Sciences.2024; 45(5): 2011. CrossRef - Insulin resistance: Risk factors, diagnostic approaches and mathematical models for clinical practice, epidemiological studies, and beyond
Janusz Krzymien, Piotr Ladyzynski
Biocybernetics and Biomedical Engineering.2024; 44(1): 55. CrossRef - Effects of silybin supplementation on growth performance, serum indexes and liver transcriptome of Peking ducks
Ziyue Zhang, Bozhi Shi, Xueze Lv, Yingchao Dong, Lei Li, Zhaofei Xia
Frontiers in Veterinary Science.2024;[Epub] CrossRef - Effects of Consuming Beverages Sweetened with Fructose, Glucose, High-Fructose Corn Syrup, Sucrose, or Aspartame on OGTT-Derived Indices of Insulin Sensitivity in Young Adults
Bettina Hieronimus, Valentina Medici, Vivien Lee, Marinelle V. Nunez, Desiree M. Sigala, Andrew A. Bremer, Chad L. Cox, Nancy L. Keim, Jean-Marc Schwarz, Giovanni Pacini, Andrea Tura, Peter J. Havel, Kimber L. Stanhope
Nutrients.2024; 16(1): 151. CrossRef - Association Between Insulin Resistance Markers and Poor Prognosis in Patients With Acute Ischemic Stroke After Intravenous Thrombolysis
Haimei Liu, Denglu Liu, Peng Zuo
The Neurologist.2024;[Epub] CrossRef - Interplay of Angiotensin Peptides, Vasopressin, and Insulin in the Heart: Experimental and Clinical Evidence of Altered Interactions in Obesity and Diabetes Mellitus
Ewa Szczepanska-Sadowska
International Journal of Molecular Sciences.2024; 25(2): 1310. CrossRef - High-Density Lipoprotein Is Located Alongside Insulin in the Islets of Langerhans of Normal and Rodent Models of Diabetes
Sahar Mohsin, Haba Elabadlah, Mariam K. Alotaiba, Suhail AlAmry, Shamma J. Almehairbi, Maha M. K. Harara, Aisha M. H. Almuhsin, Saeed Tariq, Frank Christopher Howarth, Ernest A. Adeghate
Nutrients.2024; 16(2): 313. CrossRef - Sweet triterpenoid glycoside from Cyclocarya paliurus ameliorates obesity-induced insulin resistance through inhibiting the TLR4/NF-κB/NLRP3 inflammatory pathway
Jie Li, Junyu He, Haibo He, Xiao Wang, Shuran Zhang, Yumin He, Jihong Zhang, Chengfu Yuan, HongWu Wang, Daoxiang Xu, Chaowang Pan, Huifan Yu, Kun Zou
Current Research in Food Science.2024; 8: 100677. CrossRef - Effect of supplementation with probiotics or synbiotics on cardiovascular risk factors in patients with metabolic syndrome: a systematic review and meta-analysis of randomized clinical trials
TingRui Chen, Jing Wang, ZeKun Liu, Fei Gao
Frontiers in Endocrinology.2024;[Epub] CrossRef - Diabetes mellitus progression in β-thalassaemia major patients: The impact of iron overload
Eglal Omer Mahgoub, Reem Qannita, Ayah Alalami, Ola Al Shehadat, Rabah Al Mahmoud, Ayah Dib, Alaa Al Hajji, Amani Al Hajji, Fatheya Al Khaja, Hany Dewedar, Mawieh Hamad, Jalal Taneera
Advances in Biomedical and Health Sciences.2024; 3(1): 5. CrossRef - Dietary Tomato Pectin Attenuates Hepatic Insulin Resistance and Inflammation in High-Fat-Diet Mice by Regulating the PI3K/AKT Pathway
Jing Sun, Kongyan Wu, Pan Wang, Yubin Wang, Dan Wang, Wenting Zhao, Yuanyuan Zhao, Chunhong Zhang, Xiaoyan Zhao
Foods.2024; 13(3): 444. CrossRef - Atractylodes macrocephala Koidz. and Cuscuta chinensis Lam. extract relieves insulin resistance via PI3K/Akt signalling in diabetic Drosophila
Yinghong Li, Ye Xu, Biwei Zhang, Zhigang Wang, Leilei Ma, Longyu Sun, Xiuping Wang, Yimin Lin, Ji-an Li, Chenxi Wu
Journal of Traditional and Complementary Medicine.2024;[Epub] CrossRef - Ketogenic diet ameliorates high-fat diet-induced insulin resistance in mouse skeletal muscle by alleviating endoplasmic reticulum stress
Qin Ma, Lincheng Jiang, Yuehua You, Hongbing Ni, Li Ma, Xiaojing Lin, Zhuyun Wang, Weiyan Yan, Xiaoqiu Xiao, Xinyu Li, Jibin Li
Biochemical and Biophysical Research Communications.2024; 702: 149559. CrossRef - Brain insulin resistance and Alzheimer’s disease: a systematic review
Luis Jesuino de Oliveira Andrade, Luís Matos de Oliveira, Alcina Maria Vinhaes Bittencourt, Letícia Góes de Carvalho Lourenço, Gabriela Correia Matos de Oliveira
Dementia & Neuropsychologia.2024;[Epub] CrossRef - Poorly controlled glycemia and worse beta cell function associate with higher resting and total energy expenditure in adults with obesity and type 2 diabetes: A doubly labeled water study
Kate Lillegard, John A. Del Castillo, Heidi J. Silver
Clinical Nutrition.2024; 43(3): 729. CrossRef - Gut microbiota in insulin resistance: a bibliometric analysis
Weiwei Tian, Li Liu, Ruirui Wang, Yunyun Quan, Bihua Tang, Dongmei Yu, Lei Zhang, Hua Hua, Junning Zhao
Journal of Diabetes & Metabolic Disorders.2024;[Epub] CrossRef - Association of the triglyceride glucose index with all-cause and cardiovascular mortality in a general population of Iranian adults
Ghazaal Alavi Tabatabaei, Noushin Mohammadifard, Hamed Rafiee, Fatemeh Nouri, Asieh Maghami mehr, Jamshid Najafian, Masoumeh Sadeghi, Maryam Boshtam, Hamidreza Roohafza, Fahimeh Haghighatdoost, Marzieh Taheri, Nizal Sarrafzadegan
Cardiovascular Diabetology.2024;[Epub] CrossRef - Changes in Cells Associated with Insulin Resistance
Leszek Szablewski
International Journal of Molecular Sciences.2024; 25(4): 2397. CrossRef - Evaluación de la resistencia a la insulina mediante el índice HOMA: un enfoque comparativo entre mujeres premenopáusicas y posmenopáusicas
Carlos Fernando Yauli Flores, Ericka Jazmín Tubón Luisa
Salud, Ciencia y Tecnología.2024; 4: 729. CrossRef - Insulin Resistance: The Increased Risk of Cancers
Leszek Szablewski
Current Oncology.2024; 31(2): 998. CrossRef - Muscle strength and non-alcoholic fatty liver disease/metabolic-associated fatty liver disease
Xuan-Yu Hao, Kai Zhang, Xing-Yong Huang, Fei Yang, Si-Yu Sun
World Journal of Gastroenterology.2024; 30(7): 636. CrossRef - Outcomes With Finerenone in Patients With Chronic Kidney Disease and Type 2 Diabetes by Baseline Insulin Resistance
Thomas Ebert, Stefan D. Anker, Luis M. Ruilope, Paola Fioretto, Vivian Fonseca, Guillermo E. Umpierrez, Andreas L. Birkenfeld, Robert Lawatscheck, Charlie Scott, Katja Rohwedder, Peter Rossing
Diabetes Care.2024; 47(3): 362. CrossRef - Influence of Obesity and Insulin Resistance on the Reproductive Outcome of Iraqi Women Undergoing Intracytoplasmic Sperm Injection
Sundus Ali Dawood, Hayder Ali Lafta Mossa, Mufeeda Ali Jwad
Al-Rafidain Journal of Medical Sciences ( ISSN 2789-3219 ).2024; 6(1): 179. CrossRef - Metabolic memory: mechanisms and diseases
Hao Dong, Yuezhang Sun, Lulingxiao Nie, Aimin Cui, Pengfei Zhao, Wai Keung Leung, Qi Wang
Signal Transduction and Targeted Therapy.2024;[Epub] CrossRef - Effect of preoperative oral carbohydrates on insulin resistance in patients undergoing laparoscopic cholecystectomy: a randomized controlled trial
Xiaohan Wang, Jingwen Zhuang, Jianxin Cheng, Zeyang Wang, Jingyi Sheng, Shanshan Guo, Rui Wang, Zhiping Wang
Langenbeck's Archives of Surgery.2024;[Epub] CrossRef - Beta‐Cell Tipe1 Orchestrates Insulin Secretion and Cell Proliferation by Promoting Gαs/cAMP Signaling via USP5
Lu Ding, Yang Sun, Yan Liang, Jie Zhang, Zhendong Fu, Caiyue Ren, Pengfei Li, Wen Liu, Rong Xiao, Hao Wang, Zhaoying Zhang, Xuetian Yue, Chunyang Li, Zhuanchang Wu, Yuemin Feng, Xiaohong Liang, Chunhong Ma, Lifen Gao
Advanced Science.2024;[Epub] CrossRef - Metabolic syndrome awareness in the general Korean population: results from a nationwide survey
Hyun-Jin Kim, Mi-Seung Shin, Kyung-Hee Kim, Mi-Hyang Jung, Dong-Hyuk Cho, Ju-Hee Lee, Kwang Kon Koh
The Korean Journal of Internal Medicine.2024; 39(2): 272. CrossRef - Pregnane X receptor knockout mitigates weight gain and hepatic metabolic dysregulation in female C57BL/6 J mice on a long-term high-fat diet
Lidya H. Gebreyesus, Sora Choi, Prince Neequaye, Mattia Mahmoud, Mia Mahmoud, Malvin Ofosu-Boateng, Elizabeth Twum, Daniel O. Nnamani, Lijin Wang, Nour Yadak, Sujoy Ghosh, Frank J. Gonzalez, Maxwell A. Gyamfi
Biomedicine & Pharmacotherapy.2024; 173: 116341. CrossRef - TyG-GGT is a Reliable Non-Invasive Predictor of Advanced Liver Fibrosis in Overweight or Obese Individuals
Lei Jin, Jing Gu, Zhe Zhang, Cheng-Fei Du, Fei-Qi Xu, Xiao-Kun Huang, Zhen-Yu Gao, Ying Li, Li-Li Yu, Xin Zhang, Guo-Qing Ru, Jun-Wei Liu, Lei Liang, Xiao-Dong Sun, Zun-Qiang Xiao
Obesity Surgery.2024; 34(4): 1333. CrossRef - Excess homocysteine inhibits pancreatic β-cell secretory function by repressing Zbtb20 expression
Tianqi Ding, Bo Wen, Jian Chen, Wenbin Chu, Rong Fan, Xuewei Chen
Molecular and Cellular Endocrinology.2024; 586: 112195. CrossRef - Testosterone therapy reduces insulin resistance in men with adult‐onset testosterone deficiency and metabolic syndrome. Results from the Moscow Study, a randomized controlled trial with an open‐label phase
Yuliya Tishova, Svetlana Kalinchenko, George Mskhalaya, Geoffrey Hackett, Mark Livingston, Carola König, Richard Strange, Michael Zitzmann, Amar Mann, Amro Maarouf, Sudarshan Ramachandran
Diabetes, Obesity and Metabolism.2024;[Epub] CrossRef - Preoperative contributing factors and the remission of diabetes after metabolic surgery: the mediating role of preoperative triglyceride
Lijuan Niu, Liqian Mu, Runda Wu, Shan Tong, Zhongqi Mao, Yi Yang, Jun Yin
Eating and Weight Disorders - Studies on Anorexia, Bulimia and Obesity.2024;[Epub] CrossRef - Mechanisms of Action of Potentilla discolor Bunge in Type 2 Diabetes Mellitus Based on Network Pharmacology and Experimental Verification in Drosophila
Yinghong Li, Fanwu Wu, Jianbo Zhang, Ye Xu, Hong Chang, Yueyue Yu, Chunhua Jiang, Xiujuan Gao, Huijuan Liu, Zhen Chen, Chenxi Wu, Ji-An Li
Drug Design, Development and Therapy.2024; Volume 18: 747. CrossRef - Diabetes and diabetic associative diseases: An overview of epigenetic regulations of TUG1
Mohammed Ageeli Hakami
Saudi Journal of Biological Sciences.2024; 31(5): 103976. CrossRef - Sleep quality of patients with diabetes mellitus: association with anxiety trait and state
Lidiane Bernardes Faria Vilela, Larissa Cristina dos Santos Camargos, Guilherme Rocha Rodrigues, Adelzí Auto Alves Júnior, Renato Canevari Dutra da Silva, Elton Brás Camargo Júnior
Revista Gaúcha de Enfermagem.2024;[Epub] CrossRef - Qualidade do sono de pacientes com diabetes mellitus: associação com ansiedade traço e estado
Lidiane Bernardes Faria Vilela, Larissa Cristina dos Santos Camargos, Guilherme Rocha Rodrigues, Adelzí Auto Alves Júnior, Renato Canevari Dutra da Silva, Elton Brás Camargo Júnior
Revista Gaúcha de Enfermagem.2024;[Epub] CrossRef - Association of the triglyceride-glucose index with severity of coronary stenosis and in-hospital mortality in patients with acute ST elevation myocardial infarction after percutaneous coronary intervention: a multicentre retrospective analysis cohort stud
Xin Lu, Xin Lin, Yingying Cai, Xiaoxiao Zhang, Haoyu Meng, Weiwei Chen, Peng Yu, Xiaohu Chen
BMJ Open.2024; 14(3): e081727. CrossRef - Effects of a Diabetic Microenvironment on Neurodegeneration: Special Focus on Neurological Cells
Vishal Chavda, Dhananjay Yadav, Snehal Patel, Minseok Song
Brain Sciences.2024; 14(3): 284. CrossRef - Mechanisms of body fat distribution and gluteal-femoral fat protection against metabolic disorders
Maha Alser, Khaled Naja, Mohamed A. Elrayess
Frontiers in Nutrition.2024;[Epub] CrossRef - Regulation of lipid metabolism by E3 ubiquitin ligases in lipid-associated metabolic diseases
Yuanming Zou, Ying Zhang, Mohan Li, Kexin Cao, Chunyu Song, Zhaobo Zhang, Kexin Cai, Danxi Geng, Shuxian Chen, Yanjiao Wu, Naijin Zhang, Guozhe Sun, Jing Wang, Yixiao Zhang, Yingxian Sun
International Journal of Biological Macromolecules.2024; 265: 130961. CrossRef - METS-IR and all-cause mortality in Korean over 60 years old: Korean genome and epidemiology study-health examinees (KoGES-HEXA) cohorts
Ha Eun Ryu, Dong Hyuk Jung, Seok-Jae Heo, Byoungjin Park, Yong Jae Lee
Frontiers in Endocrinology.2024;[Epub] CrossRef - Effects of vitamin family members on insulin resistance and diabetes complications
Hong-Jin Chen, Min Wang, Ding-Min Zou, Gui-You Liang, Si-Yuan Yang
World Journal of Diabetes.2024; 15(3): 568. CrossRef - Alleviative effects of the parthenolide derivative ACT001 on insulin resistance induced by sodium propionate combined with a high-fat diet and its potential mechanisms
Qian Yu, Xiang Zuo, Huijuan Bai, Shuhui Zhang, Jialu Luan, Qili Zhao, Xin Zhao, Xizeng Feng
European Journal of Pharmacology.2024; 971: 176529. CrossRef - The genetic causal relationship between type 2 diabetes, glycemic traits and venous thromboembolism, deep vein thrombosis, pulmonary embolism: a two-sample Mendelian randomization study
Mingyi Yang, Xianjie Wan, Yani Su, Ke Xu, Pengfei Wen, Binfei Zhang, Lin Liu, Zhi Yang, Peng Xu
Thrombosis Journal.2024;[Epub] CrossRef - Transgenerational inheritance of insulin resistance in offspring of white rice-fed female fruit flies
Kehinde Ahmad Adeshina, Kasimu Ghandi Ibrahim, Murtala Bello Abubakar, Mustapha Umar Imam
Scientific African.2024; 24: e02208. CrossRef - PPARβ/δ as a promising molecular drug target for liver diseases: A focused review
Xin Meng, Lin Wang, Yan-Chao Du, Dong Cheng, Tao Zeng
Clinics and Research in Hepatology and Gastroenterology.2024; 48(6): 102343. CrossRef - EPA and DHA Differentially Improve Insulin Resistance by Reducing Adipose Tissue Inflammation — Targeting GPR120/PPARγ Pathway
Xian Yang, Xudong Li, Manjiang Hu, Jie Huang, Siyan Yu, Huanting Zeng, Limei Mao
The Journal of Nutritional Biochemistry.2024; : 109648. CrossRef - The impact of diabetes mellitus on the development of psychiatric and neurological disorders
Olivia Kelly, Jillian Sullivan, Natalie Carris, Samantha Geci, Athena Martinez, Varvara Liashenko, James Colvin, Emily Misko, Gary Vanderlaan, He Liu, Prasad S. Dalvi
Brain Disorders.2024; : 100135. CrossRef - Oligonucleotide therapies for nonalcoholic steatohepatitis
Sixu Li, Feng Xiong, Songbo Zhang, Jinghua Liu, Guangping Gao, Jun Xie, Yi Wang
Molecular Therapy - Nucleic Acids.2024; 35(2): 102184. CrossRef - The Contribution of Type 2 Diabetes to Parkinson’s Disease Aetiology
Samo Ribarič
International Journal of Molecular Sciences.2024; 25(8): 4358. CrossRef - Dose-response associations of triglyceride to high-density lipoprotein cholesterol ratio and triglyceride–glucose index with arterial stiffness risk
Wenkai Zhang, Weifeng Huo, Huifang Hu, Tianze Li, Lijun Yuan, Jinli Zhang, Yifei Feng, Yuying Wu, Xueru Fu, Yamin Ke, Mengmeng Wang, Longkang Wang, Yaobing Chen, Yajuan Gao, Xi Li, Liang Sun, Jinyuan Pang, Zeqiang Zheng, Fulan Hu, Ming Zhang, Yu Liu, Dong
Lipids in Health and Disease.2024;[Epub] CrossRef - Unraveling PCOS: Exploring its causes and diagnostic challenges
Mohd Altaf Dar, Mudasir Maqbool, Zulfkar Qadrie, Irfat Ara, Afshana Qadir
Open Health.2024;[Epub] CrossRef - Prevalence of Metabolic Syndrome and Insulin Resistance in Adult Patients with Acne: Association with Traditional Cardiovascular Risk Factors and Acne Severity
Ana Cecília Arcanjo Carneiro, Jozélio Freire de Carvalho, Daniel Coelho de Sá, Carlos Ewerton Maia Rodrigues
Metabolic Syndrome and Related Disorders.2023; 21(2): 109. CrossRef - Metabolic and Genetic Association of Vitamin D with Calcium Signaling and Insulin Resistance
Najeebul Tarfeen, Khair Ul Nisa, Mir Bilal Ahmad, Ajaz Ahmad Waza, Bashir Ahmad Ganai
Indian Journal of Clinical Biochemistry.2023; 38(4): 407. CrossRef - Tinosporaside from Tinospora cordifolia Encourages Skeletal Muscle Glucose Transport through Both PI-3-Kinase- and AMPK-Dependent Mechanisms
Akansha Mishra, Khushbu Sharma, Jyotsana Pandey, Kapil Dev, Sleman Kadan, Mahendra Sahai, Ishbal Ahmad, Arvind K. Srivastava, Akhilesh K. Tamrakar, Hilal Zaid, Rakesh Maurya
Molecules.2023; 28(2): 483. CrossRef - Pathophysiology of obesity and its associated diseases
Xin Jin, Tingting Qiu, Li Li, Rilei Yu, Xiguang Chen, Changgui Li, Christopher G. Proud, Tao Jiang
Acta Pharmaceutica Sinica B.2023; 13(6): 2403. CrossRef - Hypertension, type 2 diabetes, obesity, and p53 mutations negatively correlate with metastatic colorectal cancer patients’ survival
Alessandro Ottaiano, Mariachiara Santorsola, Luisa Circelli, Francesco Perri, Marco Cascella, Francesco Sabbatino, Maurizio Capuozzo, Vincenza Granata, Silvia Zappavigna, Angela Lombardi, Marianna Scrima, Nadia Petrillo, Monica Ianniello, Marika Casillo,
Frontiers in Medicine.2023;[Epub] CrossRef - Triglyceride glucose index and poor sleep patterns in non-diabetic adults: Evidence from NHANES 2005–2016
Chi-Feng Liu, Li-Wei Chien
Frontiers in Nutrition.2023;[Epub] CrossRef - Polyphenol-Rich Extract of Fermented Chili Pepper Alleviates Insulin Resistance in HepG2 Cells via Regulating INSR, PTP1B, PPAR-γ, and AMPK Pathways
Tao Wang, Meiqi Li, Shengbao Cai, Linyan Zhou, Xiaosong Hu, Junjie Yi
Fermentation.2023; 9(2): 84. CrossRef - Effects of preoperative oral enzyme-hydrolyzed rice flour solution on gastric emptying and insulin resistance in patients undergoing laparoscopic cholecystectomy: a prospective randomized controlled trial
Yang Yuan, Guangjun Shi, Huailong Chen, Mingshan Wang, Haofei Liu, Xiao Zhang, Bin Wang, Gaofeng Zhang, Lixin Sun
BMC Anesthesiology.2023;[Epub] CrossRef - Roles of endoplasmic reticulum stress in the pathophysiology of polycystic ovary syndrome
Hiroshi Koike, Miyuki Harada, Akari Kusamoto, Zixin Xu, Tsurugi Tanaka, Nanoka Sakaguchi, Chisato Kunitomi, Jerilee M. K. Azhary, Nozomi Takahashi, Yoko Urata, Yutaka Osuga
Frontiers in Endocrinology.2023;[Epub] CrossRef - The Metabolic Score for Insulin Resistance (METS-IR) Predicts Cardiovascular Disease and Its Subtypes in Patients with Hypertension and Obstructive Sleep Apnea
Wenbo Yang, Xintian Cai, Junli Hu, Wen Wen, Heizhati Mulalibieke, Xiaoguang Yao, Ling Yao, Qing Zhu, Jing Hong, Qin Luo, Shasha Liu, Nanfang Li
Clinical Epidemiology.2023; Volume 15: 177. CrossRef - Sperm DNA damage: The possible link between obesity and male infertility, an update of the current literature
Andrew Peel, Anmol Saini, Joshua C. Deluao, Nicole O. McPherson
Andrology.2023; 11(8): 1635. CrossRef - Glucose Homeostasis, Diabetes Mellitus, and Gender-Affirming Treatment
Charalampos Milionis, Ioannis Ilias, Evangelia Venaki, Eftychia Koukkou
Biomedicines.2023; 11(3): 670. CrossRef - From Metabolic Syndrome to Type 2 Diabetes in Youth
Dario Iafusco, Roberto Franceschi, Alice Maguolo, Salvatore Guercio Nuzio, Antonino Crinò, Maurizio Delvecchio, Lorenzo Iughetti, Claudio Maffeis, Valeria Calcaterra, Melania Manco
Children.2023; 10(3): 516. CrossRef - Kcnma1 is involved in mitochondrial homeostasis in diabetes‐related skeletal muscle atrophy
Shan‐Yan Gao, Yong‐Ping Liu, Ri Wen, Xin‐Mei Huang, Ping Li, Yu‐Hang Yang, Ni Yang, Tie‐Ning Zhang
The FASEB Journal.2023;[Epub] CrossRef - Capsaicin and Zinc Signalling Pathways as Promising Targets for Managing Insulin Resistance and Type 2 Diabetes
Parisa Vahidi Ferdowsi, Kiran D. K. Ahuja, Jeffrey M. Beckett, Stephen Myers
Molecules.2023; 28(6): 2861. CrossRef - Mouse Models with SGLT2 Mutations: Toward Understanding the Role of SGLT2 beyond Glucose Reabsorption
Keiko Unno, Kyoko Taguchi, Yoshiichi Takagi, Tadashi Hase, Shinichi Meguro, Yoriyuki Nakamura
International Journal of Molecular Sciences.2023; 24(7): 6278. CrossRef - Association between triglyceride-glucose index and carotid atherosclerosis in patients with psoriatic arthritis
Wenhui Xie, Wei Bian, Zhibo Song, Xuerong Deng, Jiahao Qu, Zhuoli Zhang
Rheumatology.2023; 62(11): 3584. CrossRef - The relationship between HMGB1 and autophagy in the pathogenesis of diabetes and its complications
Kun Yang, Feng Cao, Weili Wang, Zhenyu Tian, Lu Yang
Frontiers in Endocrinology.2023;[Epub] CrossRef - Association of long-term triglyceride-glucose index level and change with the risk of cardiometabolic diseases
Wenqi Xu, Haiyan Zhao, Lishu Gao, Lu Guo, Jianrong Liu, Haixia Li, Junyan Sun, Aijun Xing, Shuohua Chen, Shouling Wu, Yuntao Wu
Frontiers in Endocrinology.2023;[Epub] CrossRef - White Adipose Tissue Dysfunction: Pathophysiology and Emergent Measurements
Natalia Santillana, Camila Astudillo-Guerrero, Amanda D’Espessailles, Gonzalo Cruz
Nutrients.2023; 15(7): 1722. CrossRef - Soy protein compared with whey protein ameliorates insulin resistance by regulating lipid metabolism, AMPK/mTOR pathway and gut microbiota in high-fat diet-fed mice
Andong Ji, Wei Chen, Chang Liu, Tianyu Zhang, Runjia Shi, Xinqi Wang, Huina Xu, Duo Li
Food & Function.2023; 14(12): 5752. CrossRef - Hepatic Insulin Resistance Model in the Male Wistar Rat Using Exogenous Insulin Glargine Administration
Victor Enrique Sarmiento-Ortega, Diana Moroni-González, Alfonso Diaz, Miguel Ángel García-González, Eduardo Brambila, Samuel Treviño
Metabolites.2023; 13(4): 572. CrossRef - Pathophysiological Effects of Contemporary Lifestyle on Evolutionary-Conserved Survival Mechanisms in Polycystic Ovary Syndrome
Jim Parker
Life.2023; 13(4): 1056. CrossRef - Metabolic dysfunction correction as a method of restoring the function of the reproductive system in women
G. E. Chernukha, V. A. Pronina
Meditsinskiy sovet = Medical Council.2023; (5): 90. CrossRef - Associations between TyG-BMI and normal-high blood pressure values and hypertension: cross-sectional evidence from a non-diabetic population
Nan Peng, Maobin Kuang, Yi Peng, Hang Yu, Shuhua Zhang, Guobo Xie, Guotai Sheng, Yang Zou
Frontiers in Cardiovascular Medicine.2023;[Epub] CrossRef - Inverse Correlation of Superoxide Dismutase and Catalase with Type 2 Diabetes among Rural Thais
Natnicha Promyos, Pornpimol Panprathip Phienluphon, Naruemon Wechjakwen, Jirayu Lainampetch, Pattaneeya Prangthip, Karunee Kwanbunjan
Nutrients.2023; 15(9): 2071. CrossRef - Interplay of skeletal muscle and adipose tissue: sarcopenic obesity
Min Jeong Park, Kyung Mook Choi
Metabolism.2023; 144: 155577. CrossRef - Oxygen-binding properties of blood in insulin resistance with different asprosin content
V.V. Zinchuk, J.S.O. Al-Jebur, N.V. Glutkina
Biomeditsinskaya Khimiya.2023; 69(2): 133. CrossRef - Associations between basal metabolic rate and insulin resistance in non-diabetic obese adults: Evidence from NHANES 2011–2018
Hai Guo, Dilihumaier Duolikun, Qiaoling Yao
International Journal of Diabetes in Developing Countries.2023; 43(6): 946. CrossRef - Biomarkers of Spinal Cord Injury in Patients Undergoing Complex Endovascular Aortic Repair Procedures—A Narrative Review of Current Literature
Anna Sotir, Johannes Klopf, Christine Brostjan, Christoph Neumayer, Wolf Eilenberg
Biomedicines.2023; 11(5): 1317. CrossRef - The Hypoglycemic Activities and Underlying Mechanisms of Two Saponins‐Rich Components from Fried Ziziphus jujuba Mill. Kernel
Yi‐Meng Li, Ke‐xin Hao, Hong Xie, Jian‐Guo Jiang
Molecular Nutrition & Food Research.2023;[Epub] CrossRef - Long-term outcomes and potential mechanisms of offspring exposed to intrauterine hyperglycemia
Yi-Shang Yan, Chun Feng, Dan-Qing Yu, Shen Tian, Yin Zhou, Yi-Ting Huang, Yi-Ting Cai, Jian Chen, Miao-Miao Zhu, Min Jin
Frontiers in Nutrition.2023;[Epub] CrossRef - The Function of MondoA and ChREBP Nutrient—Sensing Factors in Metabolic Disease
Byungyong Ahn
International Journal of Molecular Sciences.2023; 24(10): 8811. CrossRef - An Update on the Molecular and Cellular Basis of Pharmacotherapy in Type 2 Diabetes Mellitus
Mohamed Omer Mahgoub, Ifrah Ismail Ali, Jennifer O. Adeghate, Kornélia Tekes, Huba Kalász, Ernest A. Adeghate
International Journal of Molecular Sciences.2023; 24(11): 9328. CrossRef - Study on the therapeutic effect and mechanism of Tangningtongluo Tablet on diabetic mice
Zengxiaorui Cai, Xiangka Hu, Liuming Gui, Mushuang Qi, Wanjun Zhu, Ying Ren, Shuyu Yang, Chunmei Dai
Journal of Diabetes and its Complications.2023; 37(8): 108523. CrossRef - A Descriptive Review of the Action Mechanisms of Berberine, Quercetin and Silymarin on Insulin Resistance/Hyperinsulinemia and Cardiovascular Prevention
Paolo Bellavite, Serafino Fazio, Flora Affuso
Molecules.2023; 28(11): 4491. CrossRef - Relationships of neck circumference and abdominal obesity with insulin resistance considering relative handgrip strength in middle-aged and older individuals
Kayoung Lee
Archives of Gerontology and Geriatrics.2023; 114: 105097. CrossRef - Acute exposure to realistic concentrations of Bisphenol-A trigger health damage in fish: Blood parameters, gene expression, oxidative stress
Gustavo Axel Elizalde-Velázquez, Leobardo Manuel Gómez-Oliván, Selene Elizabeth Herrera-Vázquez, Karina Elisa Rosales-Pérez, Nely SanJuan-Reyes, Sandra García-Medina, Marcela Galar-Martínez
Aquatic Toxicology.2023; 261: 106610. CrossRef - Gαi‐coupled GPR41 activation increases Ca2+ influx in C2C12 cells and shows a therapeutic effect in diabetic animals
Do‐Hyung Lee, Kyung‐Sun Heo, Chang‐Seon Myung
Obesity.2023; 31(7): 1871. CrossRef - Physical inactivity induces insulin resistance in plantaris muscle through protein tyrosine phosphatase 1B activation in mice
Saori Kakehi, Yoshifumi Tamura, Shin-ichi Ikeda, Naoko Kaga, Hikari Taka, Yuya Nishida, Ryuzo Kawamori, Hirotaka Watada
Frontiers in Physiology.2023;[Epub] CrossRef - Metabolic-Associated Fatty Liver Disease and Insulin Resistance: A Review of Complex Interlinks
Thomas M. Barber, Stefan Kabisch, Andreas F. H. Pfeiffer, Martin O. Weickert
Metabolites.2023; 13(6): 757. CrossRef - Metabolic Markers Associated with Progression of Type 2 Diabetes Induced by High-Fat Diet and Single Low Dose Streptozotocin in Rats
Maria Andonova, Petko Dzhelebov, Krastina Trifonova, Penka Yonkova, Nikola Kostadinov, Krasimira Nancheva, Veselin Ivanov, Krasimira Gospodinova, Nikola Nizamov, Ilia Tsachev, Chavdar Chernev
Veterinary Sciences.2023; 10(7): 431. CrossRef - Research Progress Into Adipose Tissue Macrophages and Insulin Resistance
M Fu, L Yang, H Wang, Y Chen, X Chen, Q Hu, H Sun
Physiological Research.2023;[Epub] CrossRef - Effects of aerobic exercises in prediabetes patients: a systematic review and meta-analysis
Yifei Wang, Honglei Li, Dongxue Yang, Mengzhao Wang, Yanbai Han, Hongli Wang
Frontiers in Endocrinology.2023;[Epub] CrossRef - Associations of C-peptide with cardiometabolic parameters in women aged 25–44 years with different metabolic phenotypes
S. V. Mustafina, V. I. Alferova, L. V. Shcherbakova, E. V. Kashtanova, D. V. Denisova
Ateroscleroz.2023; 19(2): 115. CrossRef - Adipose-derived Mesenchymal Stem Cells Therapy as a new Treatment Option for Diabetes Mellitus
Agnieszka Mikłosz, Adrian Chabowski
The Journal of Clinical Endocrinology & Metabolism.2023; 108(8): 1889. CrossRef - Anthropometric Indices With Insulin Resistance in Obese Patients: A Literature Review
Khalid Khan, Anil Wanjari, Sourya Acharya, Sabiha Quazi
Cureus.2023;[Epub] CrossRef - Preventive and therapeutic effects of natural products and herbal extracts on nonalcoholic fatty liver disease/nonalcoholic steatohepatitis
Yiming Cao, Xiaoxue Fang, Mingyang Sun, Yegang Zhang, Mengyao Shan, Xintian Lan, Difu Zhu, Haoming Luo
Phytotherapy Research.2023; 37(9): 3867. CrossRef - Tuina (Chinese massage) for insulin resistance and sensitivity: A protocol for systematic review and meta-analysis of animal and human studies
Zhixuan Zhao, Jun Yan, Yuxin Ding, Yingji Wang, Yan Li, Ricardo Ney Oliveira Cobucci
PLOS ONE.2023; 18(7): e0288414. CrossRef - Rubus chingii Hu relieved the polycystic ovary syndrome with enhanced insulin sensitivity through inhibiting TXNIP/NLRP3 inflammasome signaling
Huizhen Li, Yongping Li, Ying Zhang, Li Tong, Yuping Sa, Wenping Sun
Gynecological Endocrinology.2023;[Epub] CrossRef - The Research Role of Triglyceride Glucose Index in Pre-Type 2 Diabetes
士博 徐
Advances in Clinical Medicine.2023; 13(07): 11762. CrossRef - Underlying mechanisms and molecular targets of genistein in the management of type 2 diabetes mellitus and related complications
Tao Jiang, Yuhe Dong, Wanying Zhu, Tong Wu, Linyan Chen, Yuantong Cao, Xi Yu, Ye Peng, Ling Wang, Ying Xiao, Tian Zhong
Critical Reviews in Food Science and Nutrition.2023; : 1. CrossRef - Gentiopicroside modulates glucose homeostasis in high-fat-diet and streptozotocin-induced type 2 diabetic mice
Xing Wang, Dongmei Long, Xianghong Hu, Nan Guo
Frontiers in Pharmacology.2023;[Epub] CrossRef - Review of the Case Reports on Metformin, Sulfonylurea, and Thiazolidinedione Therapies in Type 2 Diabetes Mellitus Patients
Elis Susilawati, Jutti Levita, Yasmiwar Susilawati, Sri Adi Sumiwi
Medical Sciences.2023; 11(3): 50. CrossRef - Effects of AIM2 and IFI16 on Infectious Diseases and Inflammation
Zhen Fan, Rui Chen, Wen Yin, Xiaomei Xie, Shan Wang, Chunbo Hao
Viral Immunology.2023; 36(7): 438. CrossRef - PREDICTING PROGRESSION TYPE 2 DIABETES MELLITUS: A 3-YEAR FOLLOW-UP STUDY EXAMINING RISK FACTORS FOR TYPE 2 DIABETES IN PATIENTS WITH PREDIABETES
Taras I. Griadil, Mykhaylo V. Bychko, Mykhaylo M. Hechko, Ksenia I. Chubirko, Ivan V. Chopey
Polski Merkuriusz Lekarski.2023; 51(3): 245. CrossRef - GABA Prevents Age-Related Sarcopenic Obesity in Mice with High-Fat-Diet-Induced Obesity
Heegu Jin, Hyun-Ji Oh, Boo-Yong Lee
Cells.2023; 12(17): 2146. CrossRef - Bio-Hacking Better Health—Leveraging Metabolic Biochemistry to Maximise Healthspan
Isabella D. Cooper, Yvoni Kyriakidou, Lucy Petagine, Kurtis Edwards, Bradley T. Elliott
Antioxidants.2023; 12(9): 1749. CrossRef - Hyperinsulinemia Impairs Clathrin-Mediated Endocytosis of the Insulin Receptor and Activation of Endothelial Nitric Oxide Synthase in Brain Endothelial Cells
Stephanie G. DiLucia, B. Jacob Kendrick, Catrina Sims-Robinson
International Journal of Molecular Sciences.2023; 24(19): 14670. CrossRef - Validation of a physiological type 2 diabetes model in human periodontal ligament stem cells
Dongqing Ai, Yuanyuan Yin, Xuyun Xia, Sihan Yang, Yu Sun, Jie Zhou, Han Qin, Xiaohui Xu, Jinlin Song
Oral Diseases.2023;[Epub] CrossRef - Flavonoids from Lophatherum gracile Brongn. Ameliorate Liver Damages in High-Fat Diet and Streptozotocin-Induced Diabetic Mice by Regulating PI3K/AKT and NF-Kappa B Pathways
Jian-Hua Zheng, Song-Xia Lin, Xiao-Yi Li, Chun-Yan Shen, Shao-Wei Zheng, Wen-Bin Chen, Walid Elfalleh
Journal of Food Biochemistry.2023; 2023: 1. CrossRef - Asprosin, a novel glucogenic adipokine implicated in type 2 diabetes mellitus
Hongcui Diao, Xue Li, Yeqiu Xu, Xiuli Xing, Shuguang Pang
Journal of Diabetes and its Complications.2023; 37(11): 108614. CrossRef - Impact of Fixed Combination of Metformin and Pioglitazone on Insulin Resistance of Patients with Type 2 Diabetes: Results of a Randomized Open-Label Study
Rui Sun, Lu Yuan, Yun Shen, Ziyang Shen, Bo Ding, Jianhua Ma
Diabetes, Metabolic Syndrome and Obesity.2023; Volume 16: 2911. CrossRef - Integrating network analysis and experimental validation to reveal the mechanism of pinocembrin in alleviating high glucose and free fatty acid-induced lipid accumulation in HepG2 cells
Kun Hu, Yongjin Sun, Jie Wang, Shaojun Wu, Jie Ren, Dan Su, Lidan Tang, Jinhong Gong, Hufeng Fang, Shan Xu, Hao Yang
Journal of Functional Foods.2023; 110: 105879. CrossRef - A Literature Review and a Proposed Classification of the Relationships between Ovulatory Infertility and Lifestyle Factors Based on the Three Groups of Ovulation Disorders Classified by WHO
Magdalena Skowrońska, Michał Pawłowski, Robert Milewski
Journal of Clinical Medicine.2023; 12(19): 6275. CrossRef - Targeting of insulin receptor endocytosis as a treatment to insulin resistance
Bryce Tim, Valentina L. Kouznetsova, Santosh Kesari, Igor F. Tsigelny
Journal of Diabetes and its Complications.2023; 37(11): 108615. CrossRef - A U-shaped association between the triglyceride to high-density lipoprotein cholesterol ratio and the risk of incident type 2 diabetes mellitus in Japanese men with normal glycemic levels: a population-based longitudinal cohort study
Bei Song, Kun Wang, Weilin Lu, Xiaofang Zhao, Tianci Yao, Ting Liu, Guangyu Gao, Haohui Fan, Chengyun Liu
Frontiers in Endocrinology.2023;[Epub] CrossRef - Association of triglyceride-glucose index levels with gestational diabetes mellitus in the US pregnant women: a cross-sectional study
Yan Zeng, Li Yin, Xiaoping Yin, Danqing Zhao
Frontiers in Endocrinology.2023;[Epub] CrossRef - The protective role of resveratrol in diabetic wound healing
Minglei Bi, Yonghong Qin, Lerong Wang, Jin Zhang
Phytotherapy Research.2023; 37(11): 5193. CrossRef - Correlation between alternative insulin resistance indexes and diabetic kidney disease: a retrospective study
Xiaodie Mu, Aihua Wu, Huiyue Hu, Min Yang, Hua Zhou
Endocrine.2023; 84(1): 136. CrossRef - Enhancing Muscle Intracellular Ca2+ Homeostasis and Glucose Uptake: Passive Pulsatile Shear Stress Treatment in Type 2 Diabetes
Arkady Uryash, Jordan Umlas, Alfredo Mijares, Jose A. Adams, Jose R. Lopez
Biomedicines.2023; 11(10): 2596. CrossRef - Review of Related Research on Type 2 Diabetes Related Macroangiopathy
珊珊 李
Advances in Clinical Medicine.2023; 13(09): 15001. CrossRef - Association between lipoprotein(a) and insulin resistance in Chinese adults: results from the China health and nutrition survey
Heng Wang, Jia-Li Fan
Frontiers in Endocrinology.2023;[Epub] CrossRef - Association between the Triglyceride–Glucose Index and Non-Alcoholic Fatty Liver Disease in patients with Atrial Fibrillation
Xiaozhong Li, Fenfang Zhan, Tian Peng, Zhen Xia, Juxiang Li
European Journal of Medical Research.2023;[Epub] CrossRef - Resveratrol as a potential protective compound against skeletal muscle insulin resistance
Arash Bahramzadeh, Kosar Bolandnazar, Reza Meshkani
Heliyon.2023; 9(11): e21305. CrossRef - The Potential Role of C-Reactive Protein in Metabolic-Dysfunction-Associated Fatty Liver Disease and Aging
Zheng Ding, Yuqiu Wei, Jing Peng, Siyu Wang, Guixi Chen, Jiazeng Sun
Biomedicines.2023; 11(10): 2711. CrossRef - Obesity and Bone Mineral Density Protection Paradox in Chronic Kidney Disease: Secreted Protein Acidic and Rich in Cysteine as a Piece of the Puzzle?
Abdelaziz Ghanemi, Fabrice Mac-Way
Life.2023; 13(11): 2172. CrossRef - Correlation of Lipid Profile and Apolipoprotein B/A-I Ratio with Insulin Resistance in Non-Diabetes Mellitus Subjects
Andi Heriadi Palloge, Liong Boy Kurniawan, Yuyun Widyaningsih, Husaini Umar, Nurahmi Nurahmi, Andi Alfian Zainuddin
INDONESIAN JOURNAL OF CLINICAL PATHOLOGY AND MEDICAL LABORATORY.2023; 30(1): 6. CrossRef - Endothelial progenitor cells as biomarkers of diabetes-related cardiovascular complications
Josefa Benítez-Camacho, Antonio Ballesteros, Lucía Beltrán-Camacho, Marta Rojas-Torres, Antonio Rosal-Vela, Margarita Jimenez-Palomares, Ismael Sanchez-Gomar, Mª Carmen Durán-Ruiz
Stem Cell Research & Therapy.2023;[Epub] CrossRef - Effects of ginseng peptides on the hypoglycemic activity and gut microbiota of a type 2 diabetes mellitus mice model
Caijing Han, Xiaoting Kong, Xiaohong Xia, Xinyu Huang, Zhaojie Mao, Jiaxin Han, Fuyan Shi, Yaohui Liang, Anning Wang, Fengxiang Zhang
Journal of Functional Foods.2023; 111: 105897. CrossRef - Effects of theasaponin E1 on the regulationglucose uptake of C2C12 myoblasts PI3K/Akt/mTOR signaling pathway
Ming Zhang, Zhiyun Chen, Di Tian, Zaiqiao Li, Shaning Wang, Yujie Huo, Ling Song, Juan Lu, Jun Sheng, Xu Ji, Xiao Ma
CyTA - Journal of Food.2023; 21(1): 682. CrossRef - Efficacy and underlying mechanisms of berberine against lipid metabolic diseases: a review
Yajie Cai, Qiaoning Yang, Yanqiao Yu, Furong Yang, Ruina Bai, Xiaodi Fan
Frontiers in Pharmacology.2023;[Epub] CrossRef - Adipocyte-derived exosomal miR-22-3p modulated by circadian rhythm disruption regulates insulin sensitivity in skeletal muscle cells
Haohao Zhang, Xiaoning Zhang, Saifei Wang, Lu Zheng, Hengru Guo, Yanqi Ren, Bo Qiao, Jing Wu, Di Zhao, Lijun Xu, Shengnan Ma, Xiao Hao, Yushan Yan
Journal of Biological Chemistry.2023; 299(12): 105476. CrossRef - Total Astragalus saponins can reverse type 2 diabetes mellitus-related intestinal dysbiosis and hepatic insulin resistance in vivo
Leilei Ma, Xiaojin La, Biwei Zhang, Wenxuan Xu, Chunyu Tian, Qianru Fu, Meng Wang, Chenxi Wu, Zhen Chen, Hong Chang, Ji-an Li
Frontiers in Endocrinology.2023;[Epub] CrossRef - Interplay between Vitamin D and Adipose Tissue: Implications for Adipogenesis and Adipose Tissue Function
Shiqi Lu, Zhen-Bo Cao
Nutrients.2023; 15(22): 4832. CrossRef - Alanine aminotransferase to high- density lipoprotein cholesterol ratio is positively correlated with the occurrence of diabetes in the Chinese population: a population-based cohort study
Shiming He, Changhui Yu, Maobin Kuang, Jiajun Qiu, Ruijuan Yang, Shuhua Zhang, Guotai Sheng, Yang Zou
Frontiers in Endocrinology.2023;[Epub] CrossRef - The association between the metabolic score for insulin resistance (METS-IR) index and urinary incontinence in the United States: results from the National Health and Nutrition Examination Survey (NHANES) 2001–2018
Shangqi Cao, Linghao Meng, Lede Lin, Xu Hu, Xiang Li
Diabetology & Metabolic Syndrome.2023;[Epub] CrossRef - Adipose Tissue, Non-Communicable Diseases, and Physical Exercise: An Imperfect Triangle
Francisco A. Monsalve, Fernando Delgado-López, Barbra Fernández-Tapia, Daniel R. González
International Journal of Molecular Sciences.2023; 24(24): 17168. CrossRef - Risk of metabolic abnormalities in osteoarthritis: a new perspective to understand its pathological mechanisms
Guizheng Wei, Ke Lu, Muhammad Umar, Zhenglin Zhu, William W. Lu, John R. Speakman, Yan Chen, Liping Tong, Di Chen
Bone Research.2023;[Epub] CrossRef - Influence of Sociodemographic Variables and Healthy Habits on the Values of Insulin Resistance Indicators in 386,924 Spanish Workers
Miguel Mestre Font, Carla Busquets-Cortés, José Ignacio Ramírez-Manent, Pilar Tomás-Gil, Hernán Paublini, Ángel Arturo López-González
Nutrients.2023; 15(24): 5122. CrossRef - Konjac flour-mediated gut microbiota alleviates insulin resistance and improves placental angiogenesis of obese sows
Deyuan Wu, Wenyu Xiong, Shuo Ma, Jinxi Luo, Hongxuan Ye, Shuangbo Huang, Fuyong Li, Xi’en Xiang, Qiling Chen, Binghui Gao, Jinping Deng, Yulong Yin, Chengquan Tan
AMB Express.2023;[Epub] CrossRef - Physiological Adaptations to Life in Space: An Update
Isadora de Carvalho e Silva, Thais Russomano, Ricardo Alves Ferreira, Marli do Carmo Cupertino, Fabíola Alves Alcântara, Mauro Geller, Oswaldo Monteiro Del Cima, Rodrigo Siqueira-Batista
Journal of Aerospace Technology and Management.2023;[Epub] CrossRef - Blood and Brain Metabolites after Cerebral Ischemia
Eva Baranovicova, Dagmar Kalenska, Peter Kaplan, Maria Kovalska, Zuzana Tatarkova, Jan Lehotsky
International Journal of Molecular Sciences.2023; 24(24): 17302. CrossRef - Key Therapeutic Targets to Treat Hyperglycemia-Induced Atherosclerosis Analyzed Using a Petri Net-Based Model
Agnieszka Rybarczyk, Dorota Formanowicz, Piotr Formanowicz
Metabolites.2023; 13(12): 1191. CrossRef - Exploration of the Mechanism of the Comorbidity Relationship between Alzheimer’s Disease and Diabetes Mellitus
涛 温
Medical Diagnosis.2023; 13(04): 440. CrossRef - Postbiyotikler ve İnsülin Direnci
Betül SARIDAĞ DEVRAN, Mendane SAKA
Van Sağlık Bilimleri Dergisi.2023; 16(3): 268. CrossRef - The Anti-Diabetic Potential of Baicalin: Evidence from Rodent Studies
Tomasz Szkudelski, Katarzyna Szkudelska
International Journal of Molecular Sciences.2023; 25(1): 431. CrossRef - Clinical application of Momordica charantia (Bitter Melon) for reducing blood sugar in type 2 diabetes mellitus
Ashley Dahlquist, Dana Jandali, Mirielle C. Nauman, Jeremy J. Johnson, Sasho Stoleski
International Journal of Nutrition.2023; 7(4): 8. CrossRef - Alkaloids as Promising Agents for the Management of Insulin Resistance:
A Review
Ayoub Amssayef, Mohamed Eddouks
Current Pharmaceutical Design.2023; 29(39): 3123. CrossRef - Potential Mechanisms for How Long-Term Physical Activity May Reduce Insulin Resistance
Sindre Lee-Ødegård, Thomas Olsen, Frode Norheim, Christian Andre Drevon, Kåre Inge Birkeland
Metabolites.2022; 12(3): 208. CrossRef - Pathophysiological Link between Insulin Resistance and Adrenal Incidentalomas
Jordan A. Higgs, Alyssa P. Quinn, Kevin D. Seely, Zeke Richards, Shad P. Mortensen, Cody S. Crandall, Amanda E. Brooks
International Journal of Molecular Sciences.2022; 23(8): 4340. CrossRef - The Dose-Response Effects of Consuming High Fructose Corn Syrup-Sweetened Beverages on Hepatic Lipid Content and Insulin Sensitivity in Young Adults
Desiree M. Sigala, Bettina Hieronimus, Valentina Medici, Vivien Lee, Marinelle V. Nunez, Andrew A. Bremer, Chad L. Cox, Candice A. Price, Yanet Benyam, Yasser Abdelhafez, John P. McGahan, Nancy L. Keim, Michael I. Goran, Giovanni Pacini, Andrea Tura, Clau
Nutrients.2022; 14(8): 1648. CrossRef - Association of β-cell function and cognitive impairment in patients with abnormal glucose metabolism
Mengyi Guo, Jiaokun Jia, Jia Zhang, Mingyue Zhou, Anxin Wang, Shengyun Chen, Xingquan Zhao
BMC Neurology.2022;[Epub] CrossRef - The Role of Insulin Resistance in Fueling NAFLD Pathogenesis: From Molecular Mechanisms to Clinical Implications
Rossella Palma, Annamaria Pronio, Mario Romeo, Flavia Scognamiglio, Lorenzo Ventriglia, Vittorio Maria Ormando, Antonietta Lamazza, Stefano Pontone, Alessandro Federico, Marcello Dallio
Journal of Clinical Medicine.2022; 11(13): 3649. CrossRef - Crosstalk between Schizophrenia and Metabolic Syndrome: The Role of Oxytocinergic Dysfunction
Kah Kheng Goh, Cynthia Yi-An Chen, Tzu-Hua Wu, Chun-Hsin Chen, Mong-Liang Lu
International Journal of Molecular Sciences.2022; 23(13): 7092. CrossRef - TyG index is positively associated with risk of CHD and coronary atherosclerosis severity among NAFLD patients
Jianqi Zhao, Hongxuan Fan, Ting Wang, Bing Yu, Shaobin Mao, Xun Wang, Wenjing Zhang, Leigang Wang, Yao Zhang, Zhaoyu Ren, Bin Liang
Cardiovascular Diabetology.2022;[Epub] CrossRef - The Association of Acute Phase Proteins in Stress and Inflammation-Induced T2D
Tammy Speelman, Lieke Dale, Ann Louw, Nicolette J. D. Verhoog
Cells.2022; 11(14): 2163. CrossRef - Probiotic Mechanisms Affecting Glucose Homeostasis: A Scoping Review
Maša Pintarič, Tomaž Langerholc
Life.2022; 12(8): 1187. CrossRef - Potential Molecular Targets of Oleanolic Acid in Insulin Resistance and Underlying Oxidative Stress: A Systematic Review
Ángel Fernández-Aparicio, María Correa-Rodríguez, Jose M. Castellano, Jacqueline Schmidt-RioValle, Javier S. Perona, Emilio González-Jiménez
Antioxidants.2022; 11(8): 1517. CrossRef - Impact of Highly Saturated versus Unsaturated Fat Intake on Carbohydrate Metabolism and Vascular Reactivity in Rat
Youzan Ferdinand Djohan, Fabrice Raynaud, Karen Lambert, Jean-Paul Cristol, Charles Coudray, Christine Feillet-Coudray, Anne Virsolvy, Eric Badia, Néstor Gutiérrez-Méndez
Biochemistry Research International.2022; 2022: 1. CrossRef - Mitophagy: A potential therapeutic target for insulin resistance
Peng Ning, Xiaobo Jiang, Jing Yang, Jiaxing Zhang, Fan Yang, Hongyi Cao
Frontiers in Physiology.2022;[Epub] CrossRef - Higher neutrophil to lymphocyte ratio is associated with renal dysfunction and cardiac adverse remodeling in elderly with metabolic syndrome
Yuqi Zhu, Gang Li, Jari A. Laukkanen, Xing Song, Jing Zhang, Linping Wei, Xinrui Chen, Yufeng Li, Cheng Liu
Frontiers in Cardiovascular Medicine.2022;[Epub] CrossRef - Lipid droplet accumulation in β cells in patients with type 2 diabetes is associated with insulin resistance, hyperglycemia and β cell dysfunction involving decreased insulin granules
Tomomi Horii, Junji Kozawa, Yukari Fujita, Satoshi Kawata, Harutoshi Ozawa, Chisaki Ishibashi, Sho Yoneda, Takao Nammo, Jun-ichiro Miyagawa, Hidetoshi Eguchi, Iichiro Shimomura
Frontiers in Endocrinology.2022;[Epub] CrossRef - Comparing the long non-coding RNA expression profiles of skeletal muscle and kidney tissues from patients with diabetes
Young-Kook Kim, Takahiro Nemoto
PLOS ONE.2022; 17(9): e0274794. CrossRef - The CCR2+ Monocyte Subsets Increase in Obese Boys but Not Girls with Abnormally High Carotid Intima-Media Thickness: A Pilot Study
María José Garcés-Hernández, Karen Pedraza-Escudero, Nayely Garibay-Nieto, Joselin Hernández-Ruiz, Jessica Lakshmi Prieto-Chávez, Lourdes Andrea Arriaga-Pizano, Eréndira Villanueva-Ortega, Galileo Escobedo, Aaron Noe Manjarrez-Reyna, Juan Carlos López-Alv
Journal of Cardiovascular Development and Disease.2022; 9(10): 330. CrossRef - Effect of honey bee venom on the histological changes of testes and hormonal disturbance in diabetic mice
Sattar J. J. AL-Shaeli, Talal Jabal Hussen, Ali M. Ethaeb
Veterinary World.2022; : 2357. CrossRef - Establishment and Validation of a New Predictive Model for Insulin Resistance based on 2 Chinese Cohorts: A Cross-Sectional Study
Shi Zhang, Xin-Cheng Wang, Jing Li, Xiao-He Wang, Yi Wang, Yan-Ju Zhang, Mei-Yang Du, Min-Ying Zhang, Jing-Na Lin, Chun-Jun Li, Aman Rajpal
International Journal of Endocrinology.2022; 2022: 1. CrossRef - Zishen Pill alleviates diabetes in Db/db mice via activation of PI3K/AKT pathway in the liver
You Wu, Boju Sun, Xiaoyuan Guo, Lili Wu, Yaomu Hu, Lingling Qin, Tao Yang, Mei Li, Tianyu Qin, Miao Jiang, Tonghua Liu
Chinese Medicine.2022;[Epub] CrossRef - Effect of Insulin and Pioglitazone on Protein Phosphatase 2A Interaction Partners in Primary Human Skeletal Muscle Cells Derived from Obese Insulin-Resistant Participants
Lana Alghanem, Xiangmin Zhang, Ruchi Jaiswal, Berhane Seyoum, Abdullah Mallisho, Zaher Msallaty, Zhengping Yi
ACS Omega.2022; 7(47): 42763. CrossRef - Dietary Plant Protein Intake Can Reduce Maternal Insulin Resistance during Pregnancy
Yuting Hong, Chen Yang, Jinjing Zhong, Yanmei Hou, Kui Xie, Linlin Wang
Nutrients.2022; 14(23): 5039. CrossRef - Preventive effect of probiotics supplementation on occurrence of gestational diabetes mellitus: A systematic review and meta-analysis of randomized controlled trials
Azin Pakmehr, Hanieh-Sadat Ejtahed, Nooshin Shirzad, Mahboobeh Hemmatabadi, Sara Farhat, Bagher Larijani
Frontiers in Medicine.2022;[Epub] CrossRef - Insulin resistance in ischemic stroke: Mechanisms and therapeutic approaches
Peng-Fei Ding, Hua-Sheng Zhang, Jie Wang, Yong-Yue Gao, Jian-Nan Mao, Chun-Hua Hang, Wei Li
Frontiers in Endocrinology.2022;[Epub] CrossRef - Long Noncoding RNAs in the Pathogenesis of Insulin Resistance
Weili Yang, Yixiang Lyu, Rui Xiang, Jichun Yang
International Journal of Molecular Sciences.2022; 23(24): 16054. CrossRef - Predictability of HOMA-IR for Gestational Diabetes Mellitus in Early Pregnancy Based on Different First Trimester BMI Values
Yanbei Duo, Shuoning Song, Yuemei Zhang, Xiaolin Qiao, Jiyu Xu, Jing Zhang, Zhenyao Peng, Yan Chen, Xiaorui Nie, Qiujin Sun, Xianchun Yang, Ailing Wang, Wei Sun, Yong Fu, Yingyue Dong, Zechun Lu, Tao Yuan, Weigang Zhao
Journal of Personalized Medicine.2022; 13(1): 60. CrossRef - MODERN CONCEPTS OF THE PATHOGENESIS OF TYPE 2 DIABETES MELLITUS
D.V. Kurkin, E.I. Morkovin, D.A. Bakulin, Yu.V. Gorbunova, A.V. Strygin, A.I. Robertus, I.E. Makarenko, V.B. Saparova, R.V. Drai, V.I. Petrov
Journal of Volgograd State Medical University.2022; 19(4): 34. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite