- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 37(5); 2013 > Article
-
ReviewPathophysiology Regulation of Muscle Pyruvate Dehydrogenase Complex in Insulin Resistance: Effects of Exercise and Dichloroacetate
- Dumitru Constantin-Teodosiu
-
Diabetes & Metabolism Journal 2013;37(5):301-314.
DOI: https://doi.org/10.4093/dmj.2013.37.5.301
Published online: October 17, 2013
School of Life Sciences, University of Nottingham Medical School, Nottingham, UK.
- Corresponding author: Dumitru Constantin-Teodosiu. School of Life Sciences, University of Nottingham Medical School, NG7 2UH, Nottingham, UK. tim.constantin@nottingham.ac.uk
Copyright © 2013 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- REGULATION OF PDC ACTIVITY
- MUSCLE PDC ACTIVITY AND MUSCLE ACETYLCARNITINE ACCUMULATION
- PDC AND PDK4 IN HUMAN SKELETAL MUSCLE
- INHIBITION OF MUSCLE PDC ACTIVITY BY DRUGS
- MECHANISM OF PDK4 ACTIVATION BY PPARs/FOXO1
- EFFECT OF DICHLOROACETATE ON PDC ACTIVITY AND CHO OXIDATION
- DOES THE HYPOXIA-INDUCIBLE FACTOR-1α MEDIATED PDK4 UP-REGULATION MAKE CHUVASH POLYCYTHEMIA PATIENTS INSULIN RESISTANT?
- CONCLUSIONS
- NOTES
- REFERENCES
ABSTRACT
- Since the mitochondrial pyruvate dehydrogenase complex (PDC) controls the rate of carbohydrate oxidation, impairment of PDC activity mediated by high-fat intake has been advocated as a causative factor for the skeletal muscle insulin resistance, metabolic syndrome, and the onset of type 2 diabetes (T2D). There are also situations where muscle insulin resistance can occur independently from high-fat dietary intake such as sepsis, inflammation, or drug administration though they all may share the same underlying mechanism, i.e., via activation of forkhead box family of transcription factors, and to a lower extent via peroxisome proliferator-activated receptors. The main feature of T2D is a chronic elevation in blood glucose levels. Chronic systemic hyperglycaemia is toxic and can lead to cellular dysfunction that may become irreversible over time due to deterioration of the pericyte cell's ability to provide vascular stability and control to endothelial proliferation. Therefore, it may not be surprising that T2D's complications are mainly macrovascular and microvascular related, i.e., neuropathy, retinopathy, nephropathy, coronary artery, and peripheral vascular diseases. However, life style intervention such as exercise, which is the most potent physiological activator of muscle PDC, along with pharmacological intervention such as administration of dichloroacetate or L-carnitine can prove to be viable strategies for treating muscle insulin resistance in obesity and T2D as they can potentially restore whole body glucose disposal.
- Regulation of glucose disposal by skeletal muscle and exercise
- There are globally more than 1 billion overweight adults, at least 300 million of whom are obese. The key causes of obesity are: 1) increased consumption of foods rich in saturated fats and sugars, and 2) the lack of physical activity, which leads to a rise in blood circulating free fatty acids (FFAs). A chronic elevation of circulating FFA leads to a metabolic syndrome, which is a cluster of risk factors such as insulin resistance, hypertension, and dyslipidaemia (Fig. 1). Insulin resistance can be described as the inability of skeletal muscle to switch from fat to carbohydrate (CHO) oxidation in response to a diet-mediated increase in CHO or insulin availability.
- The main function of skeletal muscle in vertebrates is the motor function. It is known that when muscle contracts (exercise) to produce force it also increases pyruvate generation by enhancing muscle glycogen degradation and leg glucose uptake from the circulating blood. Pyruvate can either be reduced to lactate or oxidised by pyruvate dehydrogenase complex (PDC) in mitochondria to produce acetyl-CoA according to the following irreversible reaction: pyruvate+coenzyme A (CoASH)+ nicotinamide adenine dinucleotide (NAD+)→ acetyl-CoA+ nicotinamide adenine dinucleotide hydrogen (NADH)+CO2. Through this reaction and its ability to increase the volume of glucose oxidised in response to exercise and insulin stimulus, skeletal muscle PDC activity (PDCa) plays an important role in the whole body glucose homeostasis.
- While 10% of the O2 from the blood circulatory system is supplied to skeletal muscle at rest, this value can increase to 80% during strenuous exercise to support the oxidative energy delivery to the contracting muscle. At the fuel substrate level, over 50% of the available glucose after a mixed-meal at rest will be stored as glycogen or oxidised by the skeletal muscles. Depending on exercise intensity the metabolic rate can increase up to 10-fold from the resting rate to meet the energy expenditure of the contracting muscle and most of energy is provided by the PDC-mediated CHO oxidation (Fig. 2) [1].
- In keeping with this observation, at the onset of submaximal muscle contraction (75% VO2max) PDCa raises rapidly, albeit at a slower rate than pyruvate formation, which may explain the initial muscle lactate accumulation, from resting values of around 0.3 to 0.5 (high value in trained subjects) to 2.0 to 3.0 (high value in trained subjects) mmol acetyl-CoA/min/kg wet muscle. Therefore, it is not unusual for muscle to oxidise 3 to 4 g of glucose per minute during the steady-state segment of a prolonged submaximal exercise [2].
- In summary, it seems that maximal food-borne glucose disposal, especially oxidatively, would require optimisation of three interrelated responsive fundamentals: muscle mass, exercise, and a suitable activation of PDC.
INTRODUCTION
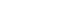
- PDC is a multicomplex mitochondrial enzyme, in fact the largest mammalian enzyme complex. PDC is composed of multiple copies of three catalytic (E1α/β-pyruvate dehydrogenase PDH, E2, and E3), one structural (E3-binding) and two regulatory proteins: PDH kinase (PDK) and PDH phosphatase (PDP). The enzyme complex is organised around a core consisting of component E2 to which E1 and E3 are joined by non-covalent bonds. Interaction of the three enzymes is brought about by lipoyl groups that visit sequentially the active sites on the three enzymes. The activation status of the PDC is controlled covalently via phosphorylation-dephosphorylation of three serine residues (Ser232, Ser293, Ser300) on the E1α subunit [3] by a competing kinase (PDK) and phosphatase (PDP) reactions [4]. The resulting interconversion cycle determines the amount of PDC existing in dephosphorylated or active form, i.e., PDCa [5]. When PDC is fully dephosphorylated the activity of PDC (maximal attainable for a given amount of protein) is labelled as total PDC (PDCt). Previous evidence from our laboratory has suggested that in humans there is a strong correlation between the amount of PDC protein and the whole body VO2max [5-7]. In other words, trained subjects have a greater maximum capacity to oxidise CHO-derived-pyruvate than sedentary subjects. Although there is no apparent explanation for this observation at a first glance, it would appear that the protein level one of the catalytic components of PDC, (E1α/β-PDH), is increased when muscle specific peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) is overexpressed [8]. Since PGC-1α seems also to control mitochondrial biogenesis and VO2max [9] then a link between PDCa and VO2max can be made.
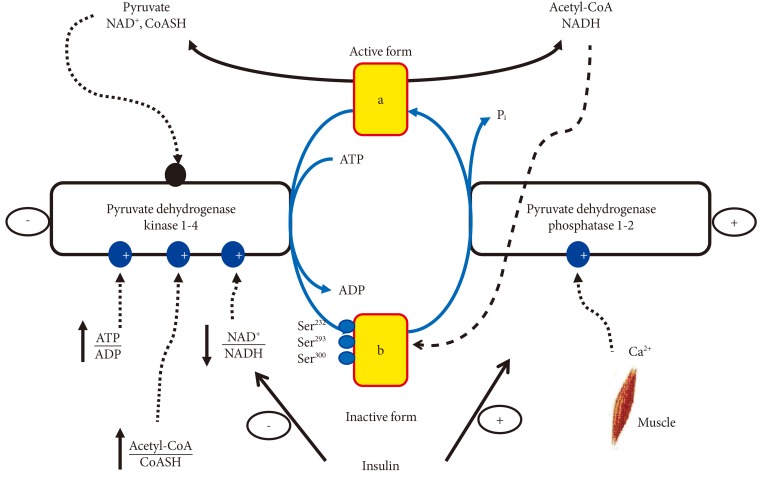
- The PDK family comprises of four isoforms (PDK1 to 4) [10], whilst PDP has two isoforms (PDP1 to 2) [11]. Although PDK2 and PDK4 mRNA are expressed in most tissues, including skeletal muscle and heart, the specific activity of PDK4 is 8-fold greater than that of PDK2 [10], thereby assigning a greater regulatory significance to PDK4. PDK1 and PDK3 appear to be limited to heart, pancreatic islets and kidney [10]. Upon muscle stimulation following a nerve-initiated electrical impulse, calcium ions (Ca2+) are released from the sarcoplasmic reticulum, which surrounds each myofibril and floods the muscle cell, which then contracts. At the same time the mitochondrial Ca2+ uptake facilitates activation of PDP1, but not PDP2 [11], and thereby dephosphorylating/activating of muscle PDC (Fig. 3).
- Of particular importance to our understanding of how muscle PDCa is regulated during muscle contraction are the early findings from in vitro studies working with purified PDC isolated from kidney, heart, and skeletal muscle, which showed that increased levels of intracellular acetyl-CoA and NADH, the FFA oxidation end products, can inhibit PDCa either by activation of PDK4 activity, and thereby covalently reducing the amount of PDCa, or by allosterically inhibiting the activity of PDCa (Fig. 3) [12,13]. Nevertheless, it is worth noting here that in vivo experiments with high intensity involuntary contraction induced by percutaneous electrical stimulation in humans showed that regardless of the contraction-induced rise in the muscle acetyl-CoA/CoASH or NADH/NAD+ ratios a complete transformation or activation of muscle PDC to PDCa occurred [7]. A similar finding was observed during voluntary submaximal exhaustive exercise where the increased muscle acetyl-CoA/CoASH ratio during exercise did not cause inhibition of either the PDC activation or the calculated catalytic activity of active PDC [5]. Collectively, these in vivo data would suggest that following a standard mixed-meal (55% to 60% CHO, 30% to 35% fat, and 10% to 15% protein) the exercise mediated muscle acetyl-CoA and NADH accumulation do not inhibit the transformation/activation of PDC to PDCa. However, allosteric inhibition of flux through a given amount of PDCa does occur by limiting the mitochondrial NAD+ and CoASH availability [14,15].
- During muscle contraction the activation of PDC is achieved by the accumulation of mitochondrial calcium and pyruvate [16], which activate PDP1 and inhibit PDK2 and 4, respectively. Jointly, this ensures that CHO utilisation and activation of PDC increase in parallel with exercise intensity (Table 1) [5,17].
- Pertinent to type 2 diabetes (T2D), however, when exercise at submaximal workloads is preceded by several days of high-fat dietary intake, calcium and pyruvate seem unable to activate PDC to the same extent as in the control diet condition [18,19], though they may at lower exercise intensities [20], resulting in reduced CHO oxidation compared to control at exercise intensities where muscle glycogen is an important contributor to energy production.
REGULATION OF PDC ACTIVITY
- It would be inappropriate to discuss exercise and activity of PDC in contracting muscle without mentioning the significance of muscle acetylcarnitine accumulation. Studies in humans have demonstrated a decrease in muscle free carnitine levels, which was matched by an almost equivalent increase in acetylcarnitine during muscle contraction [6,21]. These observed changes are consistent with the suggestion that carnitine, regulates the mitochondrial acetyl-CoA/CoASH ratio. By acting as an acceptor of acetyl groups from acetyl-CoA, carnitine may help to maintain a pool of free CoASH under conditions where the rate of acetyl-CoA condensation with oxaloacetate is less than its rate of formation from PDC mediated pyruvate decarboxylation. Thus, assuming that human muscle PDC was fully activated during exercise, it can be calculated that the rate of pyruvate decarboxylation to acetyl-CoA would be in excess of 30 µmol/sec/kg wet muscle [5]. If this rate of decarboxylation were supported solely by the CoASH available in muscle, i.e., 10 µmol/kg wet muscle [6], the entire pool of muscle CoASH would have been acetylated within one second of muscle contraction. Without the buffering effects of carnitine the PDC reaction and β-oxidation would be inhibited, since both reactions require a readily available pool of free CoASH (Fig. 4). These findings have recently prompted research into a novel approach to treating the metabolic impairment in T2D by attempting to reduce the competition for CoASH between fat and CHO oxidation by increasing muscle carnitine pool [22].
MUSCLE PDC ACTIVITY AND MUSCLE ACETYLCARNITINE ACCUMULATION
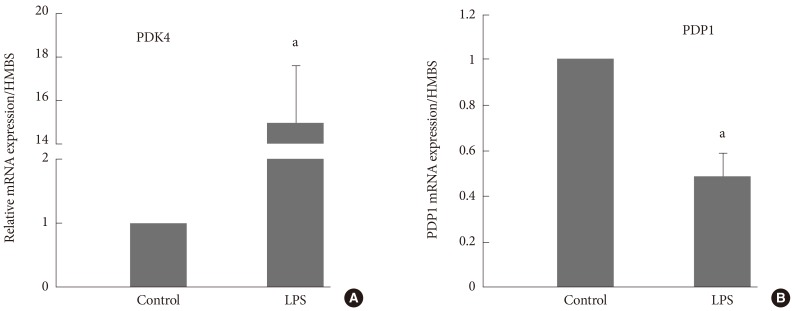
- Activity of PDC in resting muscle appears to decline whenever muscle PDK4 expression is selectively up-regulated by its main physiological regulators, i.e., changes in FFA and insulin availability, in response to starvation, hormonal and substrate changes [23-25], in pathologies such as insulin resistance and T2D [26-28], inflammation [29,30], or following medication with peroxisome proliferator-activated receptor (PPAR) agonists or statins [31,32]. T2D related muscle metabolic inflexibility is often attributed to a low level of circulating inflammatory cytokines. Indeed, in a rodent model of lipopolysaccharide (LPS) induced endotoxemia we showed that a continuous infusion of LPS over 24 hours in conscious rodents increased extensor digitorum longus muscle PDK4 mRNA expression 24-fold, PDCa decreased by 65% lower and muscle lactate accumulation increased several-fold compared with the control (saline) [30]. These changes were preceded by early marked increases in muscle tumor necrosis factor-α and interleukin-6 mRNA expression. It was concluded that the elevation in muscle lactate concentration during LPS infusion is not attributable to limited muscle oxygen availability or adenosine triphosphate (ATP) production, but rather results from inhibition of muscle PDCa, most probably due to cytokine-mediated up-regulation of PDK4 transcription. Interestingly, in another similar rodent inflammatory model it was observed that concomitant with the up-regulation of skeletal muscle PDK4, there was also a down-regulation of the muscle Ca2+-dependent PDC phosphatase (PDP1) (Fig. 5) [33].
- In T2D and starvation, states which are well associated with high circulating FFA levels, PDP1 level in heart and presumably skeletal muscle is lower than in control [34]. It is pertinent therefore to suggest that a double setback (increased PDK4 and decreased PDP1) is imposed on the muscle PDCa in T2D patients. This may potentially account for the inability of mitochondrial Ca2+/pyruvate accumulation to fully activate human muscle PDC during single bouts of submaximal intensity cycling when they were preceded by short (3 days) [2,19], or medium length (6 days) high fat dietary intake [35], when otherwise 10 minutes of exercise at 75% of VO2max, which was preceded by a normal Western diet should be sufficient to fully dephosphorylate/activate PDC [5].
- Expression of the human PDK4 gene is increased in fasting and other conditions associated with a shift from glucose utilization to fatty acids as an energy provider. However, muscle PDK4 mRNA expression can be also increased under conditions of reduced muscle glycogen availability (exercise mediated), which the authors proposed to be attributable to glycogen regulatory enzymes such as protein phosphatase 1 and glycogen synthase kinase 3, which are bound to the glycogen structure only to be released when the glycogen level decreases [36]. The authors concluded that exercise-induced expression of metabolic genes may be co-ordinately linked to signalling mechanisms sensitive to glycogen level. However, this contention along with the proposed central role of pyruvate to muscle PDC activation in vivo may be overstated, at least during muscle contraction. Thus, the importance of glycogen availability and pyruvate formation to PDC activation during intense exercise in human skeletal muscle were investigated after glycogen depleting one-legged cycling exercise [16]. During a subsequent 10 minutes of two legged-exercise at 75% VO2max, it was clear that regardless of whether pre-exercise muscle glycogen content was at a typical resting concentration or depleted, the increase in PDC activation from its resting value was the same, despite pyruvate accumulation in the glycogen depleted leg during exercise being 3-fold lower than in normal glycogen leg. However, as a result of the reduction in pyruvate availability, calculated flux through PDC reaction was several-fold lower in the glycogen depleted state compared with normal. It is therefore pertinent to conclude that whilst muscle glycogen and pyruvate availability appear to be important to the rate of flux through PDC reaction during in vivo contraction, they are not of primary importance to the control of PDC activation under these conditions, which is probably principally regulated by muscle calcium availability.
PDC AND PDK4 IN HUMAN SKELETAL MUSCLE
- After medication with PPARs agonists and statins, which were prescribed in the diabetic population to improve blood lipidemic profile by increasing fat oxidation and reducing circulation cholesterol levels, unfortunately inhibition of muscle PDCa via increase in expression of muscle PDK4 mRNA and protein can also occur. By way of illustration, in a rodent model 6 days of medication with the PPAR-δ agonist GW610742 inhibited the activity of PDC-dependent CHO oxidation (Fig. 6) [37] via up-regulation of PDK4 [31]. This impaired muscle function during a sustained contraction over 30 minutes where the demand on CHO use was markedly increased (Fig. 7).
- The importance of PDCa to the force development was also illustrated in a PDK4 knockout mice model whose extensor digitorum muscle was able to produce 40% more force than the corresponding muscle isolated from the wild type during high intensity electrically evoked contractions [38]. Since administration of PPAR agonists to humans and rodents has been linked to lower blood glucose levels, it seems therefore appropriate to suggest that any PPAR-δ mediated blood glucose lowering effect will be unlikely to be attributable to an increase in PDC-mediated oxidative glucose disposal as this route would be inhibited following PPAR-δ mediated inhibition of PDCa. Indeed, molecular and functional analyses suggest that PPAR-δ activation reduces blood glucose levels by reducing hepatic glucose output following increased glycolysis and the pentose phosphate shunt and promoting fatty acid synthesis in the liver [39].
- Statins are the most prescribed class of drugs worldwide. Statins inhibit formation of mevalonate, and thereby the rate of hepatic cholesterol biosynthesis, by inhibiting 3-hydroxy-3-methyl glutaryl CoASH reductase. Therefore, statins are used clinically for blood cholesterol reduction in hypercholesterolaemia, and their use has been associated with a reduction in mortality in patient populations with pre-existing cardiovascular diseases [40]. However, treatment with simvastatin at 80 mg/kg body weight/day over 12 days in a rodent model up-regulated gene expression consistent with increased oxidative stress and inflammation in skeletal muscle [32]. Important to the present discussion, simvastatin markedly up-regulated muscle PDK2 and PDK4 mRNAs and glycogen level, which strongly suggests that muscle glycogen oxidation was impaired. Collectively, these findings indicate instauration of a muscle insulin resistance state after simvastatin treatment. Therefore, it may not be surprising that a recent meta-analysis of five human statin studies associated intensive high dose statin therapy with increased risk of new onset T2D [41]. The authors cautiously concluded that while the benefits of taking statins outweighs the negative effects there is a need to make patients aware of this possible risk and to monitor patients for development of diabetes, especially on intensive dose therapy.
INHIBITION OF MUSCLE PDC ACTIVITY BY DRUGS
- Increases in circulating insulin level under normal dietary conditions, together with an increase in muscle Ca2+ availability during exercise appear to be the main physiological activators of muscle PDC in humans and they act by controlling the PDK4 and PDP1 activity, respectively. Activation of muscle PDC by insulin seems to occur via increased PDP1 and PDP2 activity following activation and translocation into mitochondria of the protein kinase Cδ [42].
- Conditions with high circulating FFA levels as seen after a high-fat diet or in T2D, inflammation, PPAR agonist, or statin treatment are associated with an increase in muscle PDK4 mRNA and protein expression, thereby inhibiting activity of PDC at rest and during exercise. It has previously been suggested that activation of PPAR transcription factors (PPAR-α, -δ, -γ) by ligands, such as FFAs might be a mechanism responsible for the up-regulation of muscle PDK4 mRNA expression [31,43-45]. Conversely, others have indicated that although PDK4 protein expression in an oxidative skeletal muscle is regulated by a lipid-dependent mechanism this is not obligatorily dependent on the PPAR-α signalling [46]. Secondly, the more rapid increase in PDK4 mRNA expression compared to PPAR-α mRNA expression following administration of a PPAR-α receptor agonist speaks against any involvement of PPARs in up-regulation of PDK4 [47]. Thirdly, a distinct association between levels of plasma FFA and levels of muscle PDK4 mRNA expression, together with a lack of any change in muscle PPAR-α mRNA or protein expression were documented during a 40 hours fast in humans [35]. Collectively, these data suggest other factors than PPARs could be responsible for the increase in PDK4 mRNA expression under conditions of increased circulating FFA availability.
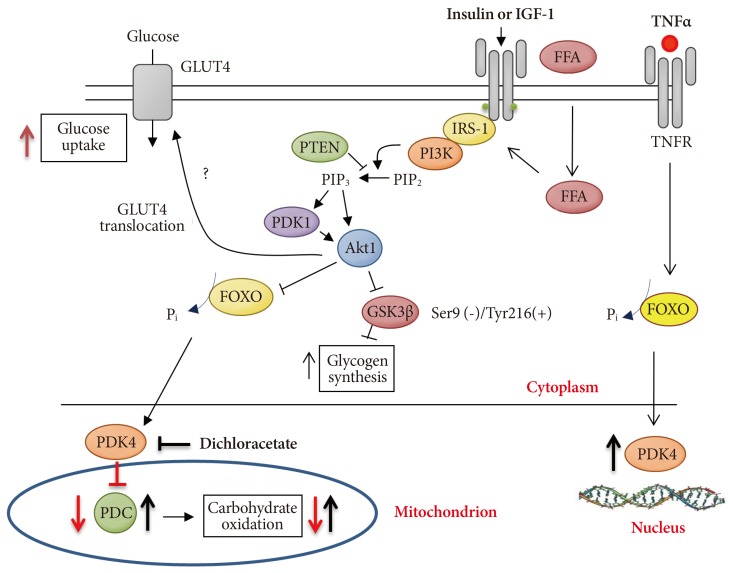
- Since FFAs can also indirectly induce the translocation of forkhead box (FOXO) transcription factors 1 and 3 to the nucleus [48,49], and FOXO1 can bind directly to the promoter region of the PDK4 gene [48], it is reasonable to suggest that FOXO factors could also play an important role in promoting the up-regulation of PDK4 mRNA in response to increased FFA availability. Indeed, the FOXO family of transcription factors have been implicated in induction of muscle insulin resistance in vivo following the upstream dysregulation of insulin mediated stimulation of phosphoinositol 3-kinase and Akt1 [50,51]. This is a pathway known to be sensitive to circulating insulin and FFA availability in humans [30]. Since FOXO1 can sense changes in availability of FFAs or insulin and relay the message downstream by modulating transcription of many skeletal muscle genes, including PDK4 [48,50], it could inhibit the PDC controlled CHO oxidation (Fig. 8).
- While these findings were documented in in vitro or in animal models [48,50], a recent study in humans in whom a muscle insulin resistant state was induced by consuming a high-fat diet for 3 days, has recognized FOXO1's involvement in up-regulation of muscle PDK4 mRNA and depression of muscle PDCa at rest and during a bout of 60 minutes of submaximal exercise [2]. Though PPAR-α mRNA was up-regulated by the high-fat diet, but to a smaller extent than PDK4 mRNA, the strong relationship between FOXO1 and PDK4 mRNA expression at rest and during exercise together with the less robust association between PPAR-α and PDK4 mRNA expression, support the notion that FOXO1 plays a more significant role than PPAR-α in the control of PDK4 expression and muscle CHO oxidation in insulin resistant state.
MECHANISM OF PDK4 ACTIVATION BY PPARs/FOXO1
- Dichloroacetate (DCA) is a halogenated carboxylic acid that has been shown to increase the activity of PDC in animal [52] and human muscles [2,53,54] by competitively inhibiting PDK2 and PDK4 [10]. Several studies have demonstrated that DCA increases CHO oxidation, reduces muscle lactate accumulation simultaneously. Consistent with this, DCA has also been shown to lower blood glucose concentration in patients with T2D [55], probably due to increased muscle and liver oxidative glucose disposal [54,56].
- Although several studies using resting [54] or contracting human muscle [53] showed that DCA infusion resulted in increased resting muscle PDCa no study, until recently, has determined whether DCA administration at rest can offset the reported high-fat diet mediated PDK2 and PDK4 inhibition of PDC activation and CHO oxidation during subsequent exercise in humans [2].
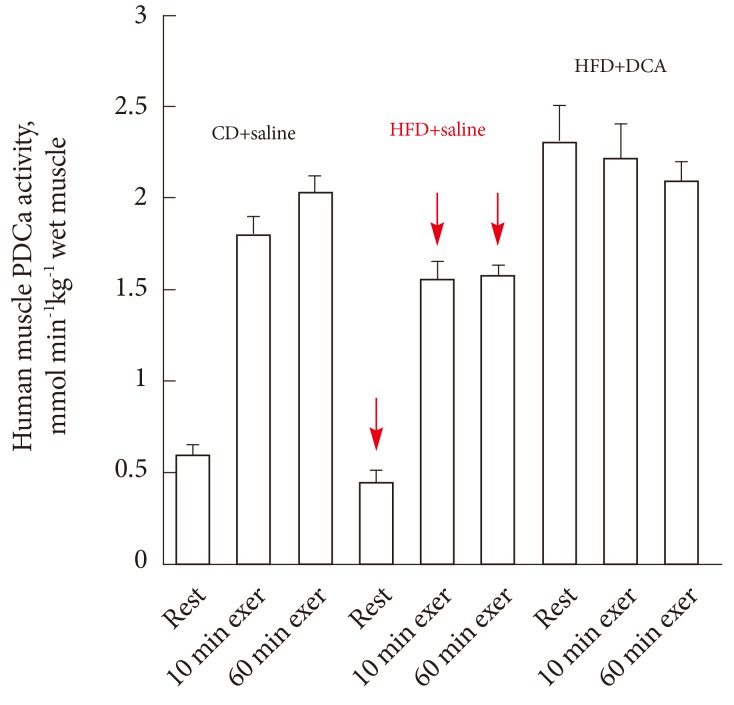
- A recent study has shown that indeed DCA administration is able to rescue the negative effects of 3-day of high-fat diet-mediated inhibition of muscle PDC activation both at rest and during subsequent exercise, and increased the rate of whole body CHO oxidation during exercise towards that seen in subjects receiving only an isocaloric control diet (Fig. 9). The authors concluded these findings support the view that PDC plays a central role in the regulation of fuel selection and insulin resistance in muscle [2].
- As discussed earlier in the present paper, administration of statins, especially at high doses, is associated with an increased prevalence of insulin resistance due to an up-regulation of muscle PDK4 isoform and decreased muscle PDCa, which collectively impair CHO oxidation. We were therefore interested to investigate whether administration of DCA could correct the impairment of CHO oxidation induced by simvastatin in a rodent model [57]. Compared with control, simvastatin (80 mg/kg/body weight) increased PDK4 protein expression and reduced muscle PDCa. However, when Simvastatin was administered with DCA (40 mg/kg/body weight) muscle PDK4 mRNA and protein were decreased while inhibition of muscle PDC activation was abolished.
- Although DCA has proved once again to be an efficient oral antidiabetic agent that could reduce blood glucose and lactate by inhibiting hepatic glucose synthesis and stimulating glucose clearance [58] and use by peripheral tissues [59], including skeletal muscle, concerns about its lack of tissue specificity and long-term safety continue to hamper its therapeutic use as an antidiabetic agent.
EFFECT OF DICHLOROACETATE ON PDC ACTIVITY AND CHO OXIDATION
- Hypoxia is a state that occurs in tissues when oxygen supplied by the cardiovascular system does not meet the demand for oxygen at cellular level. Tissue hypoxia occurs in normal physiological conditions such as contracting muscle as well as in pathophysiological settings such as inflammation [60], myocardial infarction and tumor formation [61]. In response to the low oxygen availability, the hypoxia-inducible factor (HIF) family of transcription factors are activated. One of them, HIF-1α, drives up-regulation of glycolytic enzymes to support an increase in glycolytic ATP production to bridge the decline in oxidative ATP production when mitochondrial oxygen availability is low. Experiments in cell culture have also identified a role for HIF-1α in down-regulating mitochondrial oxygen consumption by directly or indirectly inducing PDK1, which would inhibit PDCa [62]. However, these observations were restricted mainly to cell cultures or to a single PDK isoform (PDK1), and their significance for the intact organism remained largely unexplored. An opportunity to investigate the effects of altered HIF-1α levels in human skeletal muscle on all muscle PDK isoforms and PDCa was made possible by the condition of Chuvash polycythemia (CP), which is an autosomal recessive disorder with an endemic prevalence in the region of Chuvashia in the central European part of Russia. The CP patients have a mutation in the gene that controls for the von Hippel-Lindau (VHL) protein causing an arginine-to-tryptophan change at amino-acid residue 200 [63]. When bound to HIF-1α, the VHL protein allows complex recognition and subsequent destruction by the ubiquitin-proteasome proteolytic system. The change in VHL's primary amino acid structure impairs the interaction with HIF-1α, and thereby reduces the rate of HIF-α degradation and increases levels of HIF-downstream target genes under normoxic conditions. In a recent study, we measured metabolism after challenging the CP patients with a metabolic stress of exercise [64]. Exercise was a major metabolic stress for CP patients, since they had lower maximum exercise capacities and early muscle acidosis (lactate accumulation) compared with age- and gender sedentary-matched subjects. As expected for conditions characterised by high-level of HIF-1α, the skeletal muscle of the CP patients had elevated expression of glycolytic genes (i.e., hexokinase isoform 1 and 2, phosphofructokinase, M1 and M2 isoform of muscle pyruvate kinase, lactate dehydrogenase A). What was also novel was the evidence of a broad up-regulation of muscle PDK isoforms in the muscle of CP patients. Thus, three out four PDK isoforms (PDK1, 2, and 4) were markedly up-regulated compared with control. Of particular interest was PDK4 mRNA, which showed levels varying from 2- to 18-fold higher, compared to control. Recent evidence would suggest that the HIF-1α mediated up-regulation of PDK4 occurs via the orphan nuclear receptor estrogen related receptor γ [65]. Important to our understanding of why CP patients have lower exercise tolerance was the finding that the patients' muscle have lower PDP1 levels and lower muscle PDCt compared with controls. Since CP patients have high muscle PDK4 levels and low PDCt activity, if consistent with our previous discussions, they should be insulin resistant. Nevertheless, their blood glucose is lower than controls [66]. A possible explanation for this inconsistency could be that HIF-1α also promotes decreased hepatic gluconeogenesis, increased skeletal muscle glucose uptake via increased expression of GLUT1 and increased glycolysis that all contribute to a systemic decrease in blood glucose concentrations [66].
DOES THE HYPOXIA-INDUCIBLE FACTOR-1α MEDIATED PDK4 UP-REGULATION MAKE CHUVASH POLYCYTHEMIA PATIENTS INSULIN RESISTANT?
- Activation of muscle PDC is the most important tool for oxidative glucose disposal. The increase in muscle acetyl-CoA and NADH, which is normally associated with exercise does not inhibit activation of muscle PDC, but can inhibit flux through PDC pathway.
- Pyruvate availability is not important for activation of muscle PDC during exercise, but again it can limit the flux through PDC reaction. Calcium release appears to be the most important physiological activator of PDC during exercise.
- Elevated muscle PDK4 induced by dietary fat intake or inflammation can blunt both activation and the flux through PDC reaction. Acute exercise can override to some extent the above inhibition, most likely via calcium release.
- Since the response to insulin is blunted in T2D, the use of chronic/repetitive short concentric exercise (ideally high intensity training) [67], following close individual clinical control (tight monitoring of blood glucose level, the presence of silent heart ischaemia, presence of foot injuries, etc.) in combination with resistive exercise (to increase the muscle mass and therefore the whole body's ability to dispose glucose) can prove to be the norm for improving muscle insulin sensitivity (Fig. 10). A short, but intensive exercise regimen, would also not allow the glucagon to further precipitate the rise in blood glucose levels following activation of glycogenolysis and gluconeogenesis in the liver. The exercise should take place early in the morning to coincide with the dawn phenomenon, i.e., the early rise in blood glucose level.
- Alternatively, acute administration of DCA prior to exercise following several days of high-fat dietary intake can also fully activate muscle PDC and restore oxidative glucose use during exercise.
CONCLUSIONS
- 1. van Loon LJ, Greenhaff PL, Constantin-Teodosiu D, Saris WH, Wagenmakers AJ. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol 2001;536(Pt 1):295-304. ArticlePubMedPMCPDF
- 2. Constantin-Teodosiu D, Constantin D, Stephens F, Laithwaite D, Greenhaff PL. The role of FOXO and PPAR transcription factors in diet-mediated inhibition of PDC activation and carbohydrate oxidation during exercise in humans and the role of pharmacological activation of PDC in overriding these changes. Diabetes 2012;61:1017-1024. ArticlePubMedPMCPDF
- 3. Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, Dixon GH. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 1978;17:2364-2370. ArticlePubMed
- 4. Wieland OH. The mammalian pyruvate dehydrogenase complex: structure and regulation. Rev Physiol Biochem Pharmacol 1983;96:123-170. ArticlePubMed
- 5. Constantin-Teodosiu D, Cederblad G, Hultman E. PDC activity and acetyl group accumulation in skeletal muscle during prolonged exercise. J Appl Physiol 1992;73:2403-2407. ArticlePubMed
- 6. Constantin-Teodosiu D, Carlin JI, Cederblad G, Harris RC, Hultman E. Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiol Scand 1991;143:367-372. ArticlePubMed
- 7. Constantin-Teodosiu D, Cederblad G, Hultman E. PDC activity and acetyl group accumulation in skeletal muscle during isometric contraction. J Appl Physiol 1993;74:1712-1718. ArticlePubMed
- 8. Kiilerich K, Gudmundsson M, Birk JB, Lundby C, Taudorf S, Plomgaard P, Saltin B, Pedersen PA, Wojtaszewski JF, Pilegaard H. Low muscle glycogen and elevated plasma free fatty acid modify but do not prevent exercise-induced PDH activation in human skeletal muscle. Diabetes 2010;59:26-32. ArticlePubMedPDF
- 9. Tadaishi M, Miura S, Kai Y, Kano Y, Oishi Y, Ezaki O. Skeletal muscle-specific expression of PGC-1α-b, an exercise-responsive isoform, increases exercise capacity and peak oxygen uptake. PLoS One 2011;6:e28290ArticlePubMedPMC
- 10. Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-196. ArticlePubMedPMCPDF
- 11. Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM. Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem 1998;273:17680-17688. PubMed
- 12. Garland PB, Randle PJ. Control of pyruvate dehydrogenase in the perfused rat heart by the intracellular concentration of acetyl-coenzyme A. Biochem J 1964;91:6C-7C.PubMed
- 13. Tsai CS, Burgett MW, Reed LJ. Alpha-keto acid dehydrogenase complexes. XX. A kinetic study of the pyruvate dehydrogenase complex from bovine kidney. J Biol Chem 1973;248:8348-8352. PubMed
- 14. Pettit FH, Pelley JW, Reed LJ. Regulation of pyruvate dehydrogenase kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem Biophys Res Commun 1975;65:575-582. ArticlePubMed
- 15. Cooper RH, Randle PJ, Denton RM. Stimulation of phosphorylation and inactivation of pyruvate dehydrogenase by physiological inhibitors of the pyruvate dehydrogenase reaction. Nature 1975;257:808-809. ArticlePubMedPDF
- 16. Constantin-Teodosiu D, Peirce NS, Fox J, Greenhaff PL. Muscle pyruvate availability can limit the flux, but not activation, of the pyruvate dehydrogenase complex during submaximal exercise in humans. J Physiol 2004;561(Pt 2):647-655. ArticlePubMedPMC
- 17. Constantin-Teodosiu D, Cederblad G, Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Anal Biochem 1991;198:347-351. ArticlePubMed
- 18. Jansson E, Kaijser L. Effect of diet on the utilization of blood-borne and intramuscular substrates during exercise in man. Acta Physiol Scand 1982;115:19-30. ArticlePubMed
- 19. Putman CT, Spriet LL, Hultman E, Lindinger MI, Lands LC, McKelvie RS, Cederblad G, Jones NL, Heigenhauser GJ. Pyruvate dehydrogenase activity and acetyl group accumulation during exercise after different diets. Am J Physiol 1993;265(5 Pt 1):E752-E760. ArticlePubMed
- 20. St Amand TA, Spriet LL, Jones NL, Heigenhauser GJ. Pyruvate overrides inhibition of PDH during exercise after a low-carbohydrate diet. Am J Physiol Endocrinol Metab 2000;279:E275-E283. ArticlePubMed
- 21. Harris RC, Foster CV, Hultman E. Acetylcarnitine formation during intense muscular contraction in humans. J Appl Physiol 1987;63:440-442. ArticlePubMed
- 22. Stephens FB, Constantin-Teodosiu D, Greenhaff PL. New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. J Physiol 2007;581(Pt 2):431-444. ArticlePubMedPMC
- 23. Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 2002;42:249-259. ArticlePubMed
- 24. Peters SJ, Harris RA, Wu P, Pehleman TL, Heigenhauser GJ, Spriet LL. Human skeletal muscle PDH kinase activity and isoform expression during a 3-day high-fat/low-carbohydrate diet. Am J Physiol Endocrinol Metab 2001;281:E1151-E1158. ArticlePubMed
- 25. Pilegaard H, Saltin B, Neufer PD. Effect of short-term fasting and refeeding on transcriptional regulation of metabolic genes in human skeletal muscle. Diabetes 2003;52:657-662. ArticlePubMedPDF
- 26. Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes 2002;51:276-283. PubMed
- 27. Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 1999;48:1593-1599. ArticlePubMedPDF
- 28. Sugden MC, Kraus A, Harris RA, Holness MJ. Fibre-type specific modification of the activity and regulation of skeletal muscle pyruvate dehydrogenase kinase (PDK) by prolonged starvation and refeeding is associated with targeted regulation of PDK isoenzyme 4 expression. Biochem J 2000;346 Pt 3:651-657. ArticlePubMedPDF
- 29. Vary TC, Hazen S. Sepsis alters pyruvate dehydrogenase kinase activity in skeletal muscle. Mol Cell Biochem 1999;198:113-118. PubMed
- 30. Alamdari N, Constantin-Teodosiu D, Murton AJ, Gardiner SM, Bennett T, Layfield R, Greenhaff PL. Temporal changes in the involvement of pyruvate dehydrogenase complex in muscle lactate accumulation during lipopolysaccharide infusion in rats. J Physiol 2008;586:1767-1775. ArticlePubMedPMC
- 31. Constantin D, Constantin-Teodosiu D, Layfield R, Tsintzas K, Bennett AJ, Greenhaff PL. PPARdelta agonism induces a change in fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function in rat skeletal muscle. J Physiol 2007;583(Pt 1):381-390. PubMedPMC
- 32. Mallinson JE, Constantin-Teodosiu D, Sidaway J, Westwood FR, Greenhaff PL. Blunted Akt/FOXO signalling and activation of genes controlling atrophy and fuel use in statin myopathy. J Physiol 2009;587(Pt 1):219-230. ArticlePubMed
- 33. Crossland H, Constantin-Teodosiu D, Gardiner SM, Constantin D, Greenhaff PL. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J Physiol 2008;586(Pt 22):5589-5600. ArticlePubMedPMC
- 34. Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 2003;52:1371-1376. ArticlePubMedPMCPDF
- 35. Spriet LL, Tunstall RJ, Watt MJ, Mehan KA, Hargreaves M, Cameron-Smith D. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J Appl Physiol 2004;96:2082-2087. ArticlePubMed
- 36. Pilegaard H, Keller C, Steensberg A, Helge JW, Pedersen BK, Saltin B, Neufer PD. Influence of pre-exercise muscle glycogen content on exercise-induced transcriptional regulation of metabolic genes. J Physiol 2002;541(Pt 1):261-271. ArticlePubMedPMCPDF
- 37. Constantin-Teodosiu D, Baker DJ, Constantin D, Greenhaff PL. PPARdelta agonism inhibits skeletal muscle PDC activity, mitochondrial ATP production and force generation during prolonged contraction. J Physiol 2009;587(Pt 1):231-239. PubMed
- 38. Herbst EA, Dunford EC, Harris RA, Vandenboom R, Leblanc PJ, Roy BD, Jeoung NH, Peters SJ. Role of pyruvate dehydrogenase kinase 4 in regulating PDH activation during acute muscle contraction. Appl Physiol Nutr Metab 2012;37:48-52. ArticlePubMed
- 39. Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM, Evans RM. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A 2006;103:3444-3449. PubMedPMC
- 40. Vaughan CJ, Gotto AM Jr. Update on statins: 2003. Circulation 2004;110:886-892. ArticlePubMed
- 41. Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, Braunwald E, Kastelein JJ, de Lemos JA, Blazing MA, Pedersen TR, Tikkanen MJ, Sattar N, Ray KK. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA 2011;305:2556-2564. ArticlePubMed
- 42. Caruso M, Maitan MA, Bifulco G, Miele C, Vigliotta G, Oriente F, Formisano P, Beguinot F. Activation and mitochondrial translocation of protein kinase Cdelta are necessary for insulin stimulation of pyruvate dehydrogenase complex activity in muscle and liver cells. J Biol Chem 2001;276:45088-45097. PubMed
- 43. Wu P, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun 2001;287:391-396. PubMed
- 44. Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J 2005;272:3004-3014. ArticlePubMed
- 45. Degenhardt T, Saramaki A, Malinen M, Rieck M, Vaisanen S, Huotari A, Herzig KH, Muller R, Carlberg C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J Mol Biol 2007;372:341-355. PubMed
- 46. Holness MJ, Bulmer K, Gibbons GF, Sugden MC. Up-regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) protein expression in oxidative skeletal muscle does not require the obligatory participation of peroxisome-proliferator-activated receptor alpha (PPARalpha). Biochem J 2002;366(Pt 3):839-846. PubMedPMC
- 47. Motojima K. A metabolic switching hypothesis for the first step in the hypolipidemic effects of fibrates. Biol Pharm Bull 2002;25:1509-1511. ArticlePubMed
- 48. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 2003;375(Pt 2):365-371. ArticlePubMedPMCPDF
- 49. Barreyro FJ, Kobayashi S, Bronk SF, Werneburg NW, Malhi H, Gores GJ. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem 2007;282:27141-27154. ArticlePubMed
- 50. Kim YI, Lee FN, Choi WS, Lee S, Youn JH. Insulin regulation of skeletal muscle PDK4 mRNA expression is impaired in acute insulin-resistant states. Diabetes 2006;55:2311-2317. ArticlePubMedPDF
- 51. Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 2006;147:4160-4168. ArticlePubMedPDF
- 52. Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem J 1973;134:651-653. ArticlePubMedPMCPDF
- 53. Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. Am J Physiol 1998;274(2 Pt 1):E377-E380. ArticlePubMed
- 54. Constantin-Teodosiu D, Simpson EJ, Greenhaff PL. The importance of pyruvate availability to PDC activation and anaplerosis in human skeletal muscle. Am J Physiol 1999;276(3 Pt 1):E472-E478. ArticlePubMed
- 55. Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N Engl J Med 1978;298:526-530. ArticlePubMed
- 56. Shangraw RE, Rabkin JM, Lopaschuk GD. Hepatic pyruvate dehydrogenase activity in humans: effect of cirrhosis, transplantation, and dichloroacetate. Am J Physiol 1998;274(3 Pt 1):G569-G577. ArticlePubMed
- 57. Mallinson JE, Constantin-Teodosiu D, Glaves PD, Martin EA, Davies WJ, Westwood FR, Sidaway JE, Greenhaff PL. Pharmacological activation of the pyruvate dehydrogenase complex reduces statin-mediated upregulation of FOXO gene targets and protects against statin myopathy in rodents. J Physiol 2012;590(Pt 24):6389-6402. ArticlePubMedPMC
- 58. Mayers RM, Butlin RJ, Kilgour E, Leighton B, Martin D, Myatt J, Orme JP, Holloway BR. AZD7545, a novel inhibitor of pyruvate dehydrogenase kinase 2 (PDHK2), activates pyruvate dehydrogenase in vivo and improves blood glucose control in obese (fa/fa) Zucker rats. Biochem Soc Trans 2003;31(Pt 6):1165-1167. ArticlePubMedPDF
- 59. Stacpoole PW, Greene YJ. Dichloroacetate. Diabetes Care 1992;15:785-791. ArticlePubMedPDF
- 60. Shay JE, Celeste Simon M. Hypoxia-inducible factors: cross-talk between inflammation and metabolism. Semin Cell Dev Biol 2012;23:389-394. ArticlePubMed
- 61. Simon MC. Coming up for air: HIF-1 and mitochondrial oxygen consumption. Cell Metab 2006;3:150-151. ArticlePubMed
- 62. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177-185. ArticlePubMed
- 63. Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, Maxwell PH, Stockton DW, Semenza GL, Prchal JT. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet 2002;32:614-621. ArticlePubMedPDF
- 64. Formenti F, Constantin-Teodosiu D, Emmanuel Y, Cheeseman J, Dorrington KL, Edwards LM, Humphreys SM, Lappin TR, McMullin MF, McNamara CJ, Mills W, Murphy JA, O'Connor DF, Percy MJ, Ratcliffe PJ, Smith TG, Treacy M, Frayn KN, Greenhaff PL, Karpe F, Clarke K, Robbins PA. Regulation of human metabolism by hypoxia-inducible factor. Proc Natl Acad Sci U S A 2010;107:12722-12727. ArticlePubMedPMC
- 65. Lee JH, Kim EJ, Kim DK, Lee JM, Park SB, Lee IK, Harris RA, Lee MO, Choi HS. Hypoxia induces PDK4 gene expression through induction of the orphan nuclear receptor ERRγ. PLoS One 2012;7:e46324ArticlePubMedPMC
- 66. McClain DA, Abuelgasim KA, Nouraie M, Salomon-Andonie J, Niu X, Miasnikova G, Polyakova LA, Sergueeva A, Okhotin DJ, Cherqaoui R, Okhotin D, Cox JE, Swierczek S, Song J, Simon MC, Huang J, Simcox JA, Yoon D, Prchal JT, Gordeuk VR. Decreased serum glucose and glycosylated hemoglobin levels in patients with Chuvash polycythemia: a role for HIF in glucose metabolism. J Mol Med (Berl) 2013;91:59-67. ArticlePubMedPDF
- 67. Richards JC, Johnson TK, Kuzma JN, Lonac MC, Schweder MM, Voyles WF, Bell C. Short-term sprint interval training increases insulin sensitivity in healthy adults but does not affect the thermogenic response to beta-adrenergic stimulation. J Physiol 2010;588(Pt 15):2961-2972. PubMedPMC
REFERENCES

Fig. 2Energy expenditure and fuel selection during cycling exercise at 75% of VO2max. Adapted from van Loon et al. J Physiol 2001;536(Pt 1):295-304 [1]. FFA, free fatty acid.

Fig. 3Physiological factors that control muscle pyruvate dehydrogenase activity. NAD, nicotinamide adenine dinucleotide; CoASH, coenzyme A; NADH, nicotinamide adenine dinucleotide hydrogen; ATP, adenosine triphosphate; ADP, adenosine diphosphate.
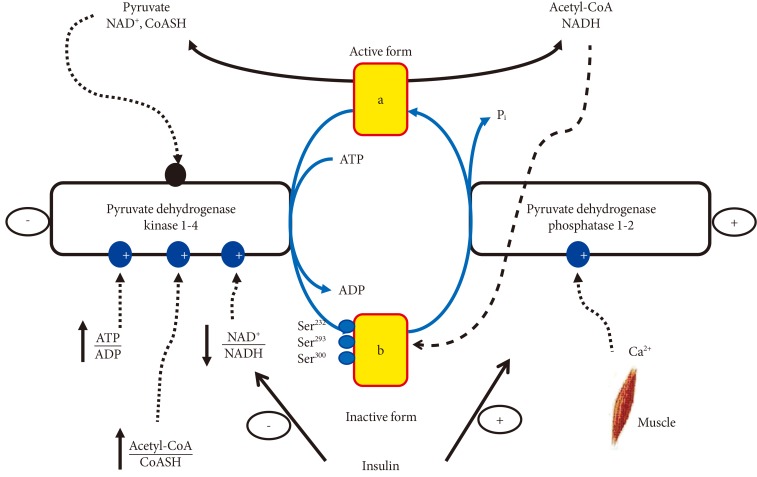
Fig. 4The competition between pyruvate dehydrogenase complex (PDC) activity-mediated carbohydrate oxidation and fat oxidation for the cellular coenzyme A (CoASH) and carnitine availability. NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide hydrogen; ATP, adenosine triphosphate; ADP, adenosine diphosphate; CPT, carnitine palmitoyl carnitine; TCA, tricarboxylic acid.
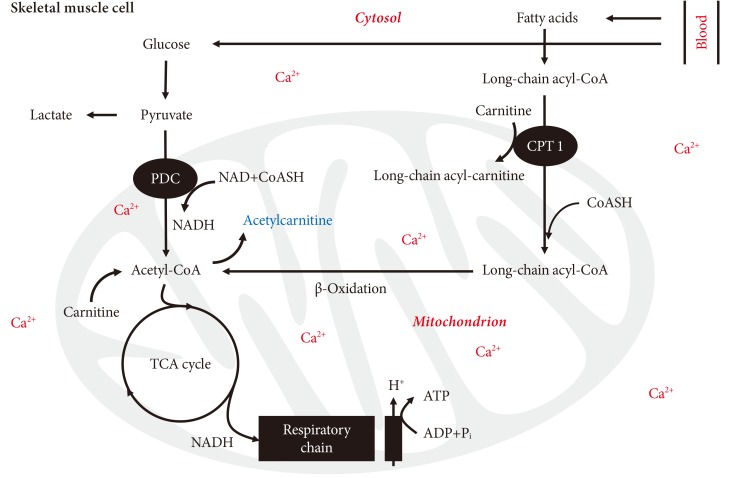
Fig. 5(A) Muscle pyruvate dehydrogenase kinase isoform 4 (PDK4) and (B) pyruvate dehydrogenase phosphatase catalytic subunit 1 (PDP1) are concomitantly up- and down-regulated, respectively, in a lipopolysaccharide (LPS)-inflammation model compared with their corresponding control (saline). Adapted from Crossland et al. J Physiol 2008;586(Pt 22):5589-600 [33]. HMBS, hydroxymethylbilane synthase. aSignificantly different from control; P<0.05.
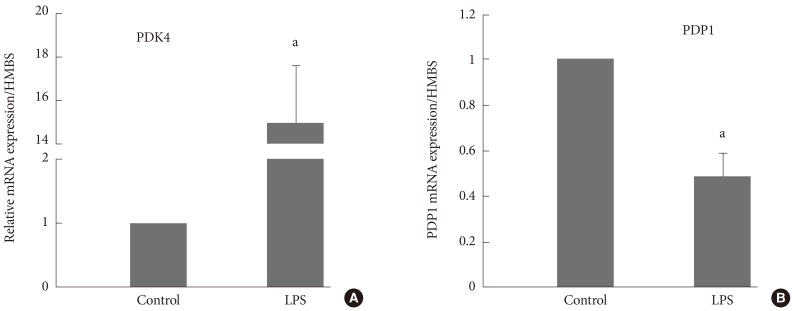
Fig. 6Muscle pyruvate dehydrogenase complex (PDC) activity at rest and after 30 minutes of electrically evoked submaximal intensity isometric contraction after 6 days of medication with different doses of peroxisome proliferator-activated receptor (PPAR)-δ agonist GW610742 (0 mg [control; empty bar], 5 mg [grey bar], and 100 mg kg-1 body weight [black bar]). Adapted from Constantin-Teodosiu et al. J Physiol 2009;587(Pt 1): 231-9 [37]. a,bSignificantly different from the corresponding control; P<0.05.
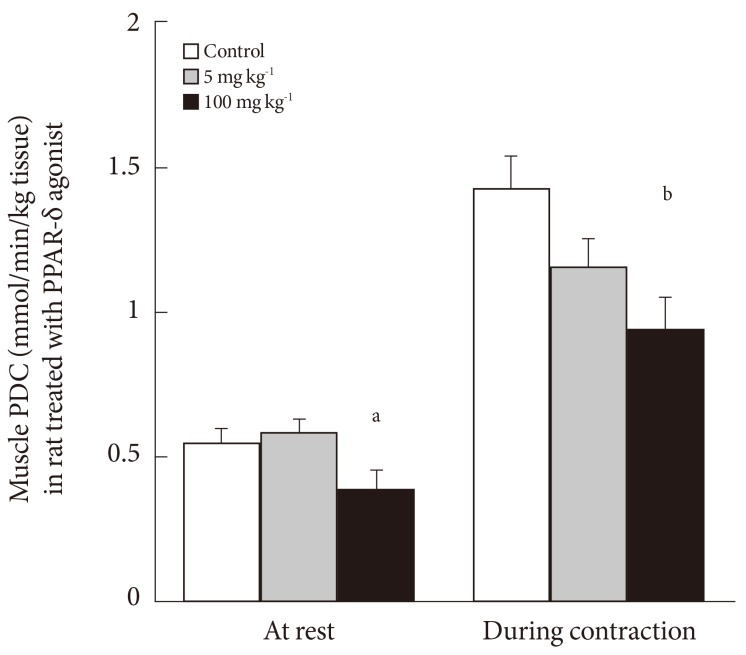
Fig. 7Rat skeletal muscle force during 30 minutes of electrically evoked submaximal intensity isometric contraction after 6 days of medication with different doses of peroxisome proliferator-activated receptor (PPAR)-δ agonist GW610742. Adapted from Constantin-Teodosiu et al. J Physiol 2009;587 (Pt 1):231-9 [37]. aSignificantly different from vehicle; P<0.05.

Fig. 8Mechanism of forkhead class O (FOXO) 1 mediated pyruvate dehydrogenase kinase isoform (PDK) 4 up-regulation and thereby pyruvate dehydrogenase complex (PDC)-mediated inhibition of carbohydrate oxidation. GLUT4, glucose transporter isoform 4; IGF, insulin-like growth factor; FFA, free fatty acid; TNF, tumor necrosis factor; PTEN, phosphatase and tensin homolog; PI3K, phosphoinositol 3-kinase; GSK, glycogen synthase; IRS-1, insulin receptor substrate 1.

Fig. 9Human quadriceps muscle pyruvate dehydrogenase complex activity (PDCa) at rest, 10 and 60 minutes of submaximal intensity exercise (75% VO2max) with dichloroacetate (DCA) or saline infusion prior to exercise after 3 days of either a standard diet (CD) or high-fat diet (HFD). Adapted from Constantin-Teodosiu et al. Diabetes 2012;61:1017-24, with permission from American Diabetes Association [2].
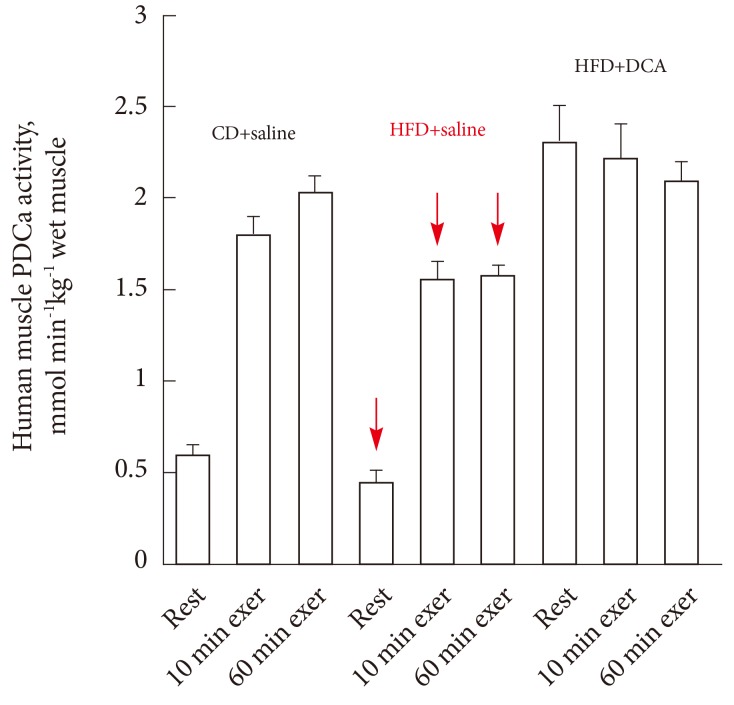
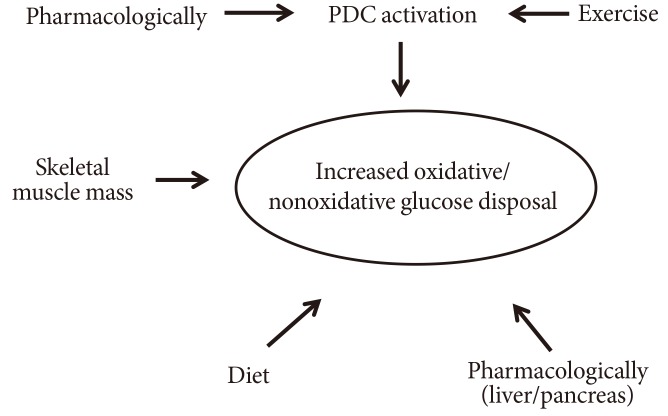
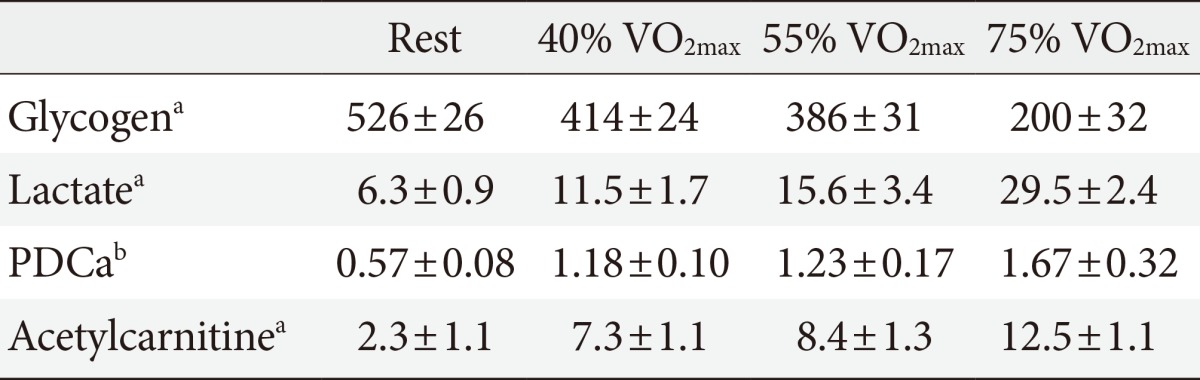
Figure & Data
References
Citations
Citations to this article as recorded by 
- Oxidative stress and metabolism meet epigenetic modulation in physical exercise
José Luis García-Giménez, Irene Cánovas-Cervera, Federico V. Pallardó
Free Radical Biology and Medicine.2024; 213: 123. CrossRef - The pyruvate dehydrogenase complex: Life’s essential, vulnerable and druggable energy homeostat
Peter W. Stacpoole, Charles E. McCall
Mitochondrion.2023; 70: 59. CrossRef - Regulating mitochondrial metabolism by targeting pyruvate dehydrogenase with dichloroacetate, a metabolic messenger
Nick Schoenmann, Nicholas Tannenbaum, Ryan M. Hodgeman, Raghavan Pillai Raju
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease.2023; 1869(7): 166769. CrossRef - Serum metabolomics profiling by proton nuclear magnetic resonance spectrometry of the response to single oral macronutrient challenges in women with polycystic ovary syndrome (PCOS) compared with male and female controls
Héctor F. Escobar-Morreale, María Ángeles Martínez-García, María Insenser, Nicolau Cañellas, Xavier Correig, Manuel Luque-Ramírez
Biology of Sex Differences.2023;[Epub] CrossRef - Metabolomics and mitochondrial dysfunction in cardiometabolic disease
Abhishek Shastry, Kimberly Dunham-Snary
Life Sciences.2023; 333: 122137. CrossRef - Loss of metabolic flexibility as a result of overexpression of pyruvate dehydrogenase kinases in muscle, liver and the immune system: Therapeutic targets in metabolic diseases
Jae‐Han Jeon, Themis Thoudam, Eun Jung Choi, Min‐Ji Kim, Robert A Harris, In‐Kyu Lee
Journal of Diabetes Investigation.2021; 12(1): 21. CrossRef - Targeting pyruvate dehydrogenase kinase signaling in the development of effective cancer therapy
Saleha Anwar, Anas Shamsi, Taj Mohammad, Asimul Islam, Md. Imtaiyaz Hassan
Biochimica et Biophysica Acta (BBA) - Reviews on Cancer.2021; 1876(1): 188568. CrossRef - Effect of exercise training on skeletal muscle protein expression in relation to insulin sensitivity: Per‐protocol analysis of a randomized controlled trial (GO‐ACTIWE)
Lea Bruhn, Rasmus Kjøbsted, Jonas Salling Quist, Anne Sofie Gram, Mads Rosenkilde, Kristine Færch, Jørgen F.P. Wojtaszewski, Bente Stallknecht, Martin Bæk Blond
Physiological Reports.2021;[Epub] CrossRef - The Mechanism behind Influenza Virus Cytokine Storm
Yinuo Gu, Xu Zuo, Siyu Zhang, Zhuoer Ouyang, Shengyu Jiang, Fang Wang, Guoqiang Wang
Viruses.2021; 13(7): 1362. CrossRef - Suppression of Pyruvate Dehydrogenase Kinase by Dichloroacetate in Cancer and Skeletal Muscle Cells Is Isoform Specific and Partially Independent of HIF-1α
Nives Škorja Milić, Klemen Dolinar, Katarina Miš, Urška Matkovič, Maruša Bizjak, Mojca Pavlin, Matej Podbregar, Sergej Pirkmajer
International Journal of Molecular Sciences.2021; 22(16): 8610. CrossRef - The Regulatory Roles of PPARs in Skeletal Muscle Fuel Metabolism and Inflammation: Impact of PPAR Agonism on Muscle in Chronic Disease, Contraction and Sepsis
Hannah Crossland, Dumitru Constantin-Teodosiu, Paul L. Greenhaff
International Journal of Molecular Sciences.2021; 22(18): 9775. CrossRef - PPARα, δ and FOXO1 Gene Silencing Overturns Palmitate-Induced Inhibition of Pyruvate Oxidation Differentially in C2C12 Myotubes
Hung-Che Chien, Despina Constantin, Paul L. Greenhaff, Dumitru Constantin-Teodosiu
Biology.2021; 10(11): 1098. CrossRef - Molecular Mechanisms of Muscle Fatigue
Dumitru Constantin-Teodosiu, Despina Constantin
International Journal of Molecular Sciences.2021; 22(21): 11587. CrossRef - Coronavirus Disease-19 (COVID-19) and Modern Lifestyle Diseases
Pallav Sengupta, Sulagna Dutta
Biomedical and Pharmacology Journal.2021; 14(4): 2245. CrossRef - A single bout of resistance exercise improves postprandial lipid metabolism in overweight/obese men with prediabetes
Adam J. Bittel, Daniel C. Bittel, Bettina Mittendorfer, Bruce W. Patterson, Adewole L. Okunade, Jun Yoshino, Lane C. Porter, Nada A. Abumrad, Dominic N. Reeds, W. Todd Cade
Diabetologia.2020; 63(3): 611. CrossRef - An integrative approach to the regulation of mitochondrial respiration during exercise: Focus on high-intensity exercise
Jose A.L. Calbet, Saúl Martín-Rodríguez, Marcos Martin-Rincon, David Morales-Alamo
Redox Biology.2020; 35: 101478. CrossRef - PPARδ and FOXO1 Mediate Palmitate-Induced Inhibition of Muscle Pyruvate Dehydrogenase Complex and CHO Oxidation, Events Reversed by Electrical Pulse Stimulation
Hung-Che Chien, Paul L. Greenhaff, Dumitru Constantin-Teodosiu
International Journal of Molecular Sciences.2020; 21(16): 5942. CrossRef - Cancer cachexia has many symptoms but only one cause: anoxia
Tomas Koltai
F1000Research.2020; 9: 250. CrossRef - Structural basis for the inhibition of PDK2 by novel ATP- and lipoyl-binding site targeting compounds
Jihoon Kang, Haushabhau S. Pagire, Donguk Kang, Yo Han Song, In Kyu Lee, Kang Taek Lee, Chin-Ju Park, Jin Hee Ahn, Jungwook Kim
Biochemical and Biophysical Research Communications.2020; 527(3): 778. CrossRef - Influenza and obesity: its odd relationship and the lessons for COVID-19 pandemic
Livio Luzi, Maria Grazia Radaelli
Acta Diabetologica.2020; 57(6): 759. CrossRef - Changes in gene expression of lactate carriers (MCT1 and CD147) in cardiac muscle of diabetic male rats: the effect of dichloroacetate and endurance training
H. Rezaeinasab, A. Habibi, M. Nikbakht, M. Rashno, S. Shakerian
The Ukrainian Biochemical Journal.2020; 92(5): 111. CrossRef - Mitochondrial Uncoupling Coordinated With PDH Activation Safely Ameliorates Hyperglycemia via Promoting Glucose Oxidation
Haowen Jiang, Jia Jin, Yanan Duan, Zhifu Xie, Yufeng Li, Anhui Gao, Min Gu, Xinwen Zhang, Chang Peng, Chunmei Xia, Tiancheng Dong, Hui Li, Lifang Yu, Jie Tang, Fan Yang, Jingya Li, Jia Li
Diabetes.2019; 68(12): 2197. CrossRef - Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Induced by Repeated Forced Swimming in Mice
Takuya Ohba, Shinichi Domoto, Miyu Tanaka, Shinsuke Nakamura, Masamitsu Shimazawa, Hideaki Hara
Biological and Pharmaceutical Bulletin.2019; 42(7): 1140. CrossRef - The Beta Cell in Type 2 Diabetes
Ashley A. Christensen, Maureen Gannon
Current Diabetes Reports.2019;[Epub] CrossRef - Reduced expression of Twist 1 is protective against insulin resistance of adipocytes and involves mitochondrial dysfunction
Sumei Lu, Hong Wang, Rui Ren, Xiaohong Shi, Yanmei Zhang, Wanshan Ma
Scientific Reports.2018;[Epub] CrossRef - PDK4 Deficiency Suppresses Hepatic Glucagon Signaling by Decreasing cAMP Levels
Bo-Yoon Park, Jae-Han Jeon, Younghoon Go, Hye Jin Ham, Jeong-Eun Kim, Eun Kyung Yoo, Woong Hee Kwon, Nam-Ho Jeoung, Yong Hyun Jeon, Seung-Hoi Koo, Byung-Gyu Kim, Ling He, Keun-Gyu Park, Robert A. Harris, In-Kyu Lee
Diabetes.2018; 67(10): 2054. CrossRef - Differences in Muscle Metabolism Between Triathletes and Normally Active Volunteers Investigated Using Multinuclear Magnetic Resonance Spectroscopy at 7T
Radka Klepochová, Ladislav Valkovič, Thomas Hochwartner, Christoph Triska, Norbert Bachl, Harald Tschan, Siegfried Trattnig, Michael Krebs, Martin Krššák
Frontiers in Physiology.2018;[Epub] CrossRef - Defining the contribution of skeletal muscle pyruvate dehydrogenase α1 to exercise performance and insulin action
Kristoffer Svensson, Jessica R. Dent, Shahriar Tahvilian, Vitor F. Martins, Abha Sathe, Julien Ochala, Mulchand S. Patel, Simon Schenk
American Journal of Physiology-Endocrinology and Metabolism.2018; 315(5): E1034. CrossRef - Early-onset and classical forms of type 2 diabetes show impaired expression of genes involved in muscle branched-chain amino acids metabolism
María Isabel Hernández-Alvarez, Angels Díaz-Ramos, María Berdasco, Jeff Cobb, Evarist Planet, Diane Cooper, Agnieszka Pazderska, Krzystof Wanic, Declan O’Hanlon, Antonio Gomez, Laura R. de la Ballina, Manel Esteller, Manuel Palacin, Donal J. O’Gorman, Joh
Scientific Reports.2017;[Epub] CrossRef - Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer
Peter W Stacpoole
JNCI: Journal of the National Cancer Institute.2017;[Epub] CrossRef - Hyperpalatable Diet and Physical Exercise Modulate the Expression of the Glial Monocarboxylate Transporters MCT1 and 4
Luis V. Portela, Andressa W. Brochier, Clarissa B. Haas, Afonso Kopczynski de Carvalho, Jussania A. Gnoato, Eduardo R. Zimmer, Eduardo Kalinine, Luc Pellerin, Alexandre P. Muller
Molecular Neurobiology.2017; 54(8): 5807. CrossRef - Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shock
Kumar Subramani, Sumin Lu, Marie Warren, Xiaogang Chu, Haroldo A. Toque, R. William Caldwell, Michael P. Diamond, Raghavan Raju
Scientific Reports.2017;[Epub] CrossRef - The effect of age and unilateral leg immobilization for 2 weeks on substrate utilization during moderate‐intensity exercise in human skeletal muscle
A. Vigelsø, M. Gram, R. Dybboe, A. B. Kuhlman, C. Prats, P. L. Greenhaff, D. Constantin‐Teodosiu, J. B. Birk, J. F. P. Wojtaszewski, F. Dela, J. W. Helge
The Journal of Physiology.2016; 594(8): 2339. CrossRef - Global Kinetic Analysis of Mammalian E3 Reveals pH-dependent NAD+/NADH Regulation, Physiological Kinetic Reversibility, and Catalytic Optimum
Michael A. Moxley, Daniel A. Beard, Jason N. Bazil
Journal of Biological Chemistry.2016; 291(6): 2712. CrossRef - Pyruvate dehydrogenase kinase regulates hepatitis C virus replication
Gwon-Soo Jung, Jae-Han Jeon, Yeon-Kyung Choi, Se Young Jang, Soo Young Park, Sung-Woo Kim, Jun-Kyu Byun, Mi-Kyung Kim, Sungwoo Lee, Eui-Cheol Shin, In-Kyu Lee, Yu Na Kang, Keun-Gyu Park
Scientific Reports.2016;[Epub] CrossRef - Altered heart proteome in fructose-fed Fisher 344 rats exposed to bisphenol A
S.A. Ljunggren, M. Iggland, M. Rönn, L. Lind, P.M. Lind, H. Karlsson
Toxicology.2016; 347-349: 6. CrossRef - Unacylated ghrelin restores insulin and autophagic signaling in skeletal muscle of diabetic mice
Bjorn T. Tam, Xiao M. Pei, Benjamin Y. Yung, Shea P. Yip, Lawrence W. Chan, Cesar S. Wong, Parco M. Siu
Pflügers Archiv - European Journal of Physiology.2015; 467(12): 2555. CrossRef - Sex-related differences in the effects of high-fat diets on DHEA-treated rats
Ana Lúcia Cecconello, Marcia Trapp, Ana Lúcia Hoefel, Cláudia Vieira Marques, Bruno Dutra Arbo, Gabriela Osterkamp, Luiz Carlos Rios Kucharski, Maria Flávia Marques Ribeiro
Endocrine.2015; 48(3): 985. CrossRef - Pharmacological Blockade of Cannabinoid CB1 Receptors in Diet-Induced Obesity Regulates Mitochondrial Dihydrolipoamide Dehydrogenase in Muscle
Sergio Arrabal, Miguel Angel Lucena, Miren Josune Canduela, Almudena Ramos-Uriarte, Patricia Rivera, Antonia Serrano, Francisco Javier Pavón, Juan Decara, Antonio Vargas, Elena Baixeras, Mercedes Martín-Rufián, Javier Márquez, Pedro Fernández-Llébrez, Bau
PLOS ONE.2015; 10(12): e0145244. CrossRef - A glance at … exercise and glucose uptake
Michael J. Glade, Kyl Smith
Nutrition.2015; 31(6): 893. CrossRef - Perpetual muscle PDH activation in PDH kinase knockout mice protects against high-fat feeding–induced muscle insulin resistance
Dumitru Constantin-Teodosiu, Francis B. Stephens, Paul L. Greenhaff
Proceedings of the National Academy of Sciences.2015;[Epub] CrossRef - Statin-Induced Increases in Atrophy Gene Expression Occur Independently of Changes in PGC1α Protein and Mitochondrial Content
Craig A. Goodman, Derk Pol, Evelyn Zacharewicz, Robert S. Lee-Young, Rod J. Snow, Aaron P. Russell, Glenn K. McConell, Ashok Kumar
PLOS ONE.2015; 10(5): e0128398. CrossRef -
13C metabolic flux analysis shows that resistin impairs the metabolic response to insulin in L6E9 myotubes
Shirley Guzmán, Silvia Marin, Anibal Miranda, Vitaly A Selivanov, Josep J Centelles, Romain Harmancey, Fatima Smih, Annie Turkieh, Yves Durocher, Antonio Zorzano, Philippe Rouet, Marta Cascante
BMC Systems Biology.2014;[Epub] CrossRef - Translational Research: From Biological Discovery to Public Benefit (or Not)
Michael R. Emmert-Buck
Advances in Biology.2014; 2014: 1. CrossRef - Diabetic Worker with History of Falls: A Case Study
Ann R. Lurati
Workplace Health & Safety.2014; 62(5): 175. CrossRef - Diabetic Worker With History of Falls: A Case Study
Ann R. Lurati
Workplace Health & Safety.2014; 62(5): 175. CrossRef - Mitochondrial Pyruvate Carrier 2 Hypomorphism in Mice Leads to Defects in Glucose-Stimulated Insulin Secretion
Patrick A. Vigueira, Kyle S. McCommis, George G. Schweitzer, Maria S. Remedi, Kari T. Chambers, Xiaorong Fu, William G. McDonald, Serena L. Cole, Jerry R. Colca, Rolf F. Kletzien, Shawn C. Burgess, Brian N. Finck
Cell Reports.2014; 7(6): 2042. CrossRef
 PubReader
PubReader Cite
Cite