- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Search
- Page Path
- HOME > Search
- Basic Research
- Extracellular Vimentin Alters Energy Metabolism And Induces Adipocyte Hypertrophy
- Ji-Hae Park, Soyeon Kwon, Young Mi Park
- Diabetes Metab J. 2024;48(2):215-230. Published online September 26, 2023
- DOI: https://doi.org/10.4093/dmj.2022.0332
- 2,279 View
- 190 Download
-
 Abstract
Abstract
 PDF
PDF Supplementary Material
Supplementary Material PubReader
PubReader  ePub
ePub - Background
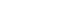
Previous studies have reported that oxidative stress contributes to obesity characterized by adipocyte hypertrophy. However, mechanism has not been studied extensively. In the current study, we evaluated role of extracellular vimentin secreted by oxidized low-density lipoprotein (oxLDL) in energy metabolism in adipocytes.
Methods
We treated 3T3-L1-derived adipocytes with oxLDL and measured vimentin which was secreted in the media. We evaluated changes in uptake of glucose and free fatty acid, expression of molecules functioning in energy metabolism, synthesis of adenosine triphosphate (ATP) and lactate, markers for endoplasmic reticulum (ER) stress and autophagy in adipocytes treated with recombinant vimentin.
Results
Adipocytes secreted vimentin in response to oxLDL. Microscopic evaluation revealed that vimentin treatment induced increase in adipocyte size and increase in sizes of intracellular lipid droplets with increased intracellular triglyceride. Adipocytes treated with vimentin showed increased uptake of glucose and free fatty acid with increased expression of plasma membrane glucose transporter type 1 (GLUT1), GLUT4, and CD36. Vimentin treatment increased transcription of GLUT1 and hypoxia-inducible factor 1α (Hif-1α) but decreased GLUT4 transcription. Adipose triglyceride lipase (ATGL), peroxisome proliferator-activated receptor γ (PPARγ), sterol regulatory element-binding protein 1 (SREBP1), diacylglycerol O-acyltransferase 1 (DGAT1) and 2 were decreased by vimentin treatment. Markers for ER stress were increased and autophagy was impaired in vimentin-treated adipocytes. No change was observed in synthesis of ATP and lactate in the adipocytes treated with vimentin.
Conclusion
We concluded that extracellular vimentin regulates expression of molecules in energy metabolism and promotes adipocyte hypertrophy. Our results show that vimentin functions in the interplay between oxidative stress and metabolism, suggesting a mechanism by which adipocyte hypertrophy is induced in oxidative stress.
- Basic Research
- Role of SUMO-Specific Protease 2 in Leptin-Induced Fatty Acid Metabolism in White Adipocytes
- Praise Chanmee Kim, Ji Seon Lee, Sung Soo Chung, Kyong Soo Park
- Diabetes Metab J. 2023;47(3):382-393. Published online March 6, 2023
- DOI: https://doi.org/10.4093/dmj.2022.0156
- 3,194 View
- 158 Download
- 1 Web of Science
- 1 Crossref
-
Abstract
PDFSupplementary MaterialPubReader ePub
- Background
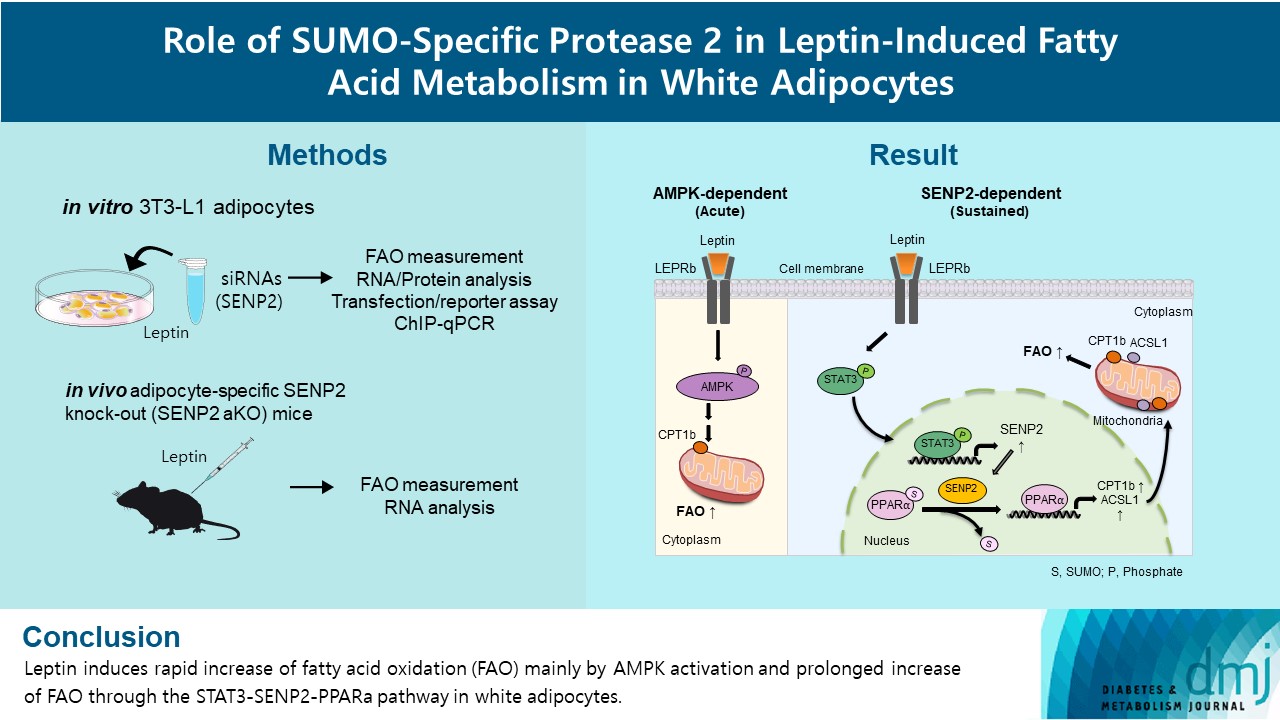
Leptin is a 16-kDa fat-derived hormone with a primary role in controlling adipose tissue levels. Leptin increases fatty acid oxidation (FAO) acutely through adenosine monophosphate-activated protein kinase (AMPK) and on delay through the SUMO-specific protease 2 (SENP2)–peroxisome proliferator-activated receptor δ/γ (PPARδ/γ) pathway in skeletal muscle. Leptin also directly increases FAO and decreases lipogenesis in adipocytes; however, the mechanism behind these effects remains unknown. Here, we investigated the role of SENP2 in the regulation of fatty acid metabolism by leptin in adipocytes and white adipose tissues.
Methods
The effects of leptin mediated by SENP2 on fatty acid metabolism were tested by siRNA-mediated knockdown in 3T3-L1 adipocytes. The role of SENP2 was confirmed in vivo using adipocyte-specific Senp2 knockout (Senp2-aKO) mice. We revealed the molecular mechanism involved in the leptin-induced transcriptional regulation of carnitine palmitoyl transferase 1b (Cpt1b) and long-chain acyl-coenzyme A synthetase 1 (Acsl1) using transfection/reporter assays and chromatin immunoprecipitation.
Results
SENP2 mediated the increased expression of FAO-associated enzymes, CPT1b and ACSL1, which peaked 24 hours after leptin treatment in adipocytes. In contrast, leptin stimulated FAO through AMPK during the initial several hours after treatment. In white adipose tissues, FAO and mRNA levels of Cpt1b and Acsl1 were increased by 2-fold 24 hours after leptin injection in control mice but not in Senp2-aKO mice. Leptin increased PPARα binding to the Cpt1b and Acsl1 promoters in adipocytes through SENP2.
Conclusion
These results suggest that the SENP2-PPARα pathway plays an important role in leptin-induced FAO in white adipocytes. -
Citations
Citations to this article as recorded by
- Intermittent cold stimulation affects energy metabolism and improves stress resistance in broiler heart
Tingting Li, Haidong Wei, Shijie Zhang, Xiaotao Liu, Lu Xing, Yuanyuan Liu, Rixin Gong, Jianhong Li
Poultry Science.2024; 103(1): 103190. CrossRef
- Intermittent cold stimulation affects energy metabolism and improves stress resistance in broiler heart
- Basic Research
- Brown Fat as a Regulator of Systemic Metabolism beyond Thermogenesis
- Okamatsu-Ogura Yuko, Masayuki Saito
- Diabetes Metab J. 2021;45(6):840-852. Published online June 25, 2021
- DOI: https://doi.org/10.4093/dmj.2020.0291

- 9,100 View
- 507 Download
- 17 Crossref
-
 Graphical Abstract
Abstract
PDFPubReader ePub
Graphical Abstract
Abstract
PDFPubReader ePub - Brown adipose tissue (BAT) is a specialized tissue for nonshivering thermogenesis to dissipate energy as heat. Although BAT research has long been limited mostly in small rodents, the rediscovery of metabolically active BAT in adult humans has dramatically promoted the translational studies on BAT in health and diseases. Moreover, several remarkable advancements have been made in brown fat biology over the past decade: The molecular and functional analyses of inducible thermogenic adipocytes (socalled beige adipocytes) arising from a developmentally different lineage from classical brown adipocytes have been accelerated. In addition to a well-established thermogenic activity of uncoupling protein 1 (UCP1), several alternative thermogenic mechanisms have been discovered, particularly in beige adipocytes. It has become clear that BAT influences other peripheral tissues and controls their functions and systemic homeostasis of energy and metabolic substrates, suggesting BAT as a metabolic regulator, other than for thermogenesis. This notion is supported by discovering that various paracrine and endocrine factors are secreted from BAT. We review the current understanding of BAT pathophysiology, particularly focusing on its role as a metabolic regulator in small rodents and also in humans.
-
Citations
Citations to this article as recorded by- Brown adipose tissue evaluation using water and triglyceride as indices by diffuse reflectance spectroscopy
Tomomi Iida, Yukio Ueda, Hideo Tsukada, Dai Fukumoto, Takafumi Hamaoka
Journal of Biophotonics.2024;[Epub] CrossRef - White-brown adipose tissue interplay in polycystic ovary syndrome: Therapeutic avenues
Khadijeh Abbasi, Reza Zarezadeh, Amir Valizadeh, Amir Mehdizadeh, Hamed Hamishehkar, Mohammad Nouri, Masoud Darabi
Biochemical Pharmacology.2024; 220: 116012. CrossRef - Brown Adipose Tissue, Batokines, and Bioactive Compounds in Foods: An Update
Fabiane Ferreira Martins, Bruna Cadete Martins, Ananda Vitoria Silva Teixeira, Matheus Ajackson, Vanessa Souza‐Mello, Julio Beltrame Daleprane
Molecular Nutrition & Food Research.2024;[Epub] CrossRef - Plasticity of Adipose Tissues: Interconversion among White, Brown, and Beige Fat and Its Role in Energy Homeostasis
Yanqiu Peng, Lixia Zhao, Min Li, Yunfei Liu, Yuke Shi, Jian Zhang
Biomolecules.2024; 14(4): 483. CrossRef - Homotaurine exhibits contrasting effects of DRD1-mediated thermogenesis-related regulators in C2C12 myoblasts and 3T3−L1 white adipocytes
Kiros Haddish, Jong Won Yun
Biotechnology and Bioprocess Engineering.2024;[Epub] CrossRef - Thermogenic Brown Fat in Humans: Implications in Energy Homeostasis, Obesity and Metabolic Disorders
Masayuki Saito, Yuko Okamatsu-Ogura
The World Journal of Men's Health.2023; 41(3): 489. CrossRef - Interplay of skeletal muscle and adipose tissue: sarcopenic obesity
Min Jeong Park, Kyung Mook Choi
Metabolism.2023; 144: 155577. CrossRef - White adipose tissue undergoes browning during preweaning period in association with microbiota formation in mice
Anju Tsukada, Yuko Okamatsu-Ogura, Emi Futagawa, Yuki Habu, Natsumi Takahashi, Mira Kato-Suzuki, Yuko Kato, Satoshi Ishizuka, Kei Sonoyama, Kazuhiro Kimura
iScience.2023; 26(7): 107239. CrossRef - In situ fluorescence-photoacoustic measurement of the changes of brown adipose tissue in mice under hindlimb unloading
Baojie Gong, Jianxin Tang, Xiaoxiao Jiang, Zhe Zhang, Shiying Li, Hongjun Jin, Liming Nie, Guojia Huang
Journal of Applied Physiology.2023; 135(2): 251. CrossRef - Age-Related Expression Dynamics of Uncoupling Protein 1 in Adipose Tissues of ICR Outbred Mice during Postnatal Ontogenesis
A. V. Yakunenkov, E. I. Elsukova, I. O. Natochy
Journal of Evolutionary Biochemistry and Physiology.2023; 59(4): 1020. CrossRef - UNCOUPLING PROTEIN UCP1 EXPRESSION DYNAMICS IN ADIPOSE TISSUES OF THE OUTBRED ICR MICE IN POSTNATAL ONTOGENESIS
A. V. Yakunenkov, E. I. Elsukova, I. O. Natochy
Журнал эволюционной биохимии и физиологии.2023; 59(4): 255. CrossRef - Antibodies Regulate Dual-Function Enzyme IYD to Induce Functional Synergy between Metabolism and Thermogenesis
Sunghyun Kang, Hwan-Woo Park, Kyung Ho Han
International Journal of Molecular Sciences.2022; 23(14): 7834. CrossRef - Machine learning-featured Secretogranin V is a circulating diagnostic biomarker for pancreatic adenocarcinomas associated with adipopenia
Yunju Jo, Min-Kyung Yeo, Tam Dao, Jeongho Kwon, Hyon‐Seung Yi, Dongryeol Ryu
Frontiers in Oncology.2022;[Epub] CrossRef - Possible roles of exercise and apelin against pregnancy complications
Hamed Alizadeh Pahlavani
Frontiers in Endocrinology.2022;[Epub] CrossRef - Relationships between the expression of adipose genes and profiles of hospitalized dogs
Yukina Sugiyama, Fumie Shimokawa, Kazutoshi Sugiyama, Takashi Kobayashi, Yusuke Yamashita, Kei Kazama, Ken Onda, Masayuki Funaba, Masaru Murakami
Veterinary Research Communications.2022; 46(4): 1239. CrossRef - Garlic (Allium sativum L.) in diabetes and its complications: Recent advances in mechanisms of action
Yayi Jiang, Rensong Yue, Guojie Liu, Jun Liu, Bo Peng, Maoyi Yang, Lianxue Zhao, Zihan Li
Critical Reviews in Food Science and Nutrition.2022; : 1. CrossRef - Fruit of Gardenia jasminoides Induces Mitochondrial Activation and Non-Shivering Thermogenesis through Regulation of PPARγ
Woo Yong Park, Gahee Song, Ja Yeon Park, Kwan-Il Kim, Kwang Seok Ahn, Hyun Jeong Kwak, Jungtae Leem, Jae-Young Um, Jinbong Park
Antioxidants.2021; 10(9): 1418. CrossRef
- Brown adipose tissue evaluation using water and triglyceride as indices by diffuse reflectance spectroscopy
- Pathophysiology
- Mitochondrial Dysfunction in Adipocytes as a Primary Cause of Adipose Tissue Inflammation
- Chang-Yun Woo, Jung Eun Jang, Seung Eun Lee, Eun Hee Koh, Ki-Up Lee
- Diabetes Metab J. 2019;43(3):247-256. Published online March 27, 2019
- DOI: https://doi.org/10.4093/dmj.2018.0221
- 8,546 View
- 257 Download
- 69 Web of Science
- 70 Crossref
-
Abstract
PDFPubReader
Adipose tissue inflammation is considered a major contributing factor in the development of obesity-associated insulin resistance and cardiovascular diseases. However, the cause of adipose tissue inflammation is presently unclear. The role of mitochondria in white adipocytes has long been neglected because of their low abundance. However, recent evidence suggests that mitochondria are essential for maintaining metabolic homeostasis in white adipocytes. In a series of recent studies, we found that mitochondrial function in white adipocytes is essential to the synthesis of adiponectin, which is the most abundant adipokine synthesized from adipocytes, with many favorable effects on metabolism, including improvement of insulin sensitivity and reduction of atherosclerotic processes and systemic inflammation. From these results, we propose a new hypothesis that mitochondrial dysfunction in adipocytes is a primary cause of adipose tissue inflammation and compared this hypothesis with a prevailing concept that “adipose tissue hypoxia” may underlie adipose tissue dysfunction in obesity. Recent studies have emphasized the role of the mitochondrial quality control mechanism in maintaining mitochondrial function. Future studies are warranted to test whether an inadequate mitochondrial quality control mechanism is responsible for mitochondrial dysfunction in adipocytes and adipose tissue inflammation.
-
Citations
Citations to this article as recorded by- Prolonged Endurance Exercise Increases Macrophage Content and Mitochondrial Respiration in Adipose Tissue in Trained Men
Ronni Eg Sahl, Ioanna Patsi, Mikkel Thunestvedt Hansen, Tue Rømer, Jacob Frandsen, Hanne Kruuse Rasmusen, Arthur Ingersen, Steen Seier Poulsen, Flemming Dela, Steen Larsen, Jørn Wulff Helge
The Journal of Clinical Endocrinology & Metabolism.2024; 109(2): e799. CrossRef - Diabetes Mellitus, Energy Metabolism, and COVID-19
Caterina Conte, Elisa Cipponeri, Michael Roden
Endocrine Reviews.2024; 45(2): 281. CrossRef - The Role of Ion-Transporting Proteins in Human Disease
Yoshinori Marunaka
International Journal of Molecular Sciences.2024; 25(3): 1726. CrossRef - The Role of Obesity in Type 2 Diabetes Mellitus—An Overview
Preethi Chandrasekaran, Ralf Weiskirchen
International Journal of Molecular Sciences.2024; 25(3): 1882. CrossRef - The Metabolic Syndrome, a Human Disease
Marià Alemany
International Journal of Molecular Sciences.2024; 25(4): 2251. CrossRef - Inflammation‐mediated metabolic regulation in adipose tissue
Shujie Xu, Feng Lu, Jianhua Gao, Yi Yuan
Obesity Reviews.2024;[Epub] CrossRef - Sleeve Gastrectomy Reduces Oxidative Stress and Reverses Mitochondrial Dysfunction Associated with Metabolic Syndrome
Micaela M. Rossi, Franco J. Signorini, Tomas A. Castillo, María P. Scribano Parada, Federico Moser, Maria dC Baez
Obesity Surgery.2024;[Epub] CrossRef - Could very low-calorie ketogenic diets turn off low grade inflammation in obesity? Emerging evidence
Luigi Barrea, Massimiliano Caprio, Mikiko Watanabe, Giuseppe Cammarata, Alessandra Feraco, Giovanna Muscogiuri, Ludovica Verde, Annamaria Colao, Silvia Savastano
Critical Reviews in Food Science and Nutrition.2023; 63(26): 8320. CrossRef - The emergent role of mitochondrial RNA modifications in metabolic alterations
Hatim Boughanem, Yvonne Böttcher, João Tomé‐Carneiro, María‐Carmen López de las Hazas, Alberto Dávalos, Akin Cayir, Manuel Macias‐González
WIREs RNA.2023;[Epub] CrossRef - Age‐associated adipose tissue inflammation promotes monocyte chemotaxis and enhances atherosclerosis
Jianrui Song, Diana Farris, Paola Ariza, Smriti Moorjani, Mita Varghese, Muriel Blin, Judy Chen, Daniel Tyrrell, Min Zhang, Kanakadurga Singer, Morgan Salmon, Daniel R. Goldstein
Aging Cell.2023;[Epub] CrossRef - Obesity, diabetes mellitus, and cardiometabolic risk: An Obesity Medicine Association (OMA) Clinical Practice Statement (CPS) 2023
Harold Edward Bays, Shagun Bindlish, Tiffany Lowe Clayton
Obesity Pillars.2023; 5: 100056. CrossRef - A role of STING signaling in obesity-induced lung inflammation
Yong Qi, Zhuhua Wu, Dan Chen, Li Zhu, Yunlei Yang
International Journal of Obesity.2023; 47(4): 325. CrossRef - Estrogens in Adipose Tissue Physiology and Obesity-Related Dysfunction
Alina Kuryłowicz
Biomedicines.2023; 11(3): 690. CrossRef - White Adipose Tissue Dysfunction: Pathophysiology and Emergent Measurements
Natalia Santillana, Camila Astudillo-Guerrero, Amanda D’Espessailles, Gonzalo Cruz
Nutrients.2023; 15(7): 1722. CrossRef - Pleiotropic and multi-systemic actions of physical exercise on PGC-1α signaling during the aging process
Ivo Vieira de Sousa Neto, Ana Paula Pinto, Vitor Rosetto Muñoz, Rita de Cássia Marqueti, José Rodrigo Pauli, Eduardo Rochete Ropelle, Adelino Sanchez Ramos da Silva
Ageing Research Reviews.2023; 87: 101935. CrossRef - The impact of metabolic endotoxaemia on the browning process in human adipocytes
Farah Omran, Alice M. Murphy, Awais Z. Younis, Ioannis Kyrou, Jana Vrbikova, Vojtech Hainer, Petra Sramkova, Martin Fried, Graham Ball, Gyanendra Tripathi, Sudhesh Kumar, Philip G. McTernan, Mark Christian
BMC Medicine.2023;[Epub] CrossRef - Molecular Mechanisms of Obesity-Induced Development of Insulin Resistance and Promotion of Amyloid-β Accumulation: Dietary Therapy Using Weak Organic Acids via Improvement of Lowered Interstitial Fluid pH
Yoshinori Marunaka
Biomolecules.2023; 13(5): 779. CrossRef - From Obesity-Induced Low-Grade Inflammation to Lipotoxicity and Mitochondrial Dysfunction: Altered Multi-Crosstalk between Adipose Tissue and Metabolically Active Organs
Gina Cavaliere, Fabiano Cimmino, Giovanna Trinchese, Angela Catapano, Lidia Petrella, Margherita D’Angelo, Lucio Lucchin, Maria Pina Mollica
Antioxidants.2023; 12(6): 1172. CrossRef - Receptor for the Advanced Glycation End Products (RAGE) Pathway in Adipose Tissue Metabolism
Klaudia Gutowska, Krzysztof Czajkowski, Alina Kuryłowicz
International Journal of Molecular Sciences.2023; 24(13): 10982. CrossRef - Aerobic and Resistance Training Attenuate Differently Knee Joint Damage Caused by a High-Fat–High-Sucrose Diet in a Rat Model
Nada Abughazaleh, Kevin Boldt, Jaqueline Lourdes Rios, Stela Marcia Mattiello, Kelsey H. Collins, Ruth-Anne Seerattan, Walter Herzog
CARTILAGE.2023;[Epub] CrossRef - Exercise mitigates age-related metabolic diseases by improving mitochondrial dysfunction
Dandan Jia, Zhenjun Tian, Ru Wang
Ageing Research Reviews.2023; 91: 102087. CrossRef - Mitochondrial dynamics and metabolism across skin cells: implications for skin homeostasis and aging
Ines Martic, Federica Papaccio, Barbara Bellei, Maria Cavinato
Frontiers in Physiology.2023;[Epub] CrossRef - Influence of Breastfeeding on the State of Meta-Inflammation in Obesity—A Narrative Review
Dominika Mazur, Małgorzata Satora, Anna K. Rekowska, Zuzanna Kabała, Aleksandra Łomża, Żaneta Kimber-Trojnar, Bożena Leszczyńska-Gorzelak
Current Issues in Molecular Biology.2023; 45(11): 9003. CrossRef - AGER-1 Long Non-Coding RNA Levels Correlate with the Expression of the Advanced Glycosylation End-Product Receptor, a Regulator of the Inflammatory Response in Visceral Adipose Tissue of Women with Obesity and Type 2 Diabetes Mellitus
Klaudia Gutowska, Krzysztof Koźniewski, Michał Wąsowski, Marta Izabela Jonas, Zbigniew Bartoszewicz, Wojciech Lisik, Maurycy Jonas, Artur Binda, Paweł Jaworski, Wiesław Tarnowski, Bartłomiej Noszczyk, Monika Puzianowska-Kuźnicka, Krzysztof Czajkowski, Ali
International Journal of Molecular Sciences.2023; 24(24): 17447. CrossRef - Pharmacological treatment with FGF21 strongly improves plasma cholesterol metabolism to reduce atherosclerosis
Cong Liu, Milena Schönke, Enchen Zhou, Zhuang Li, Sander Kooijman, Mariëtte R Boon, Mikael Larsson, Kristina Wallenius, Niek Dekker, Louise Barlind, Xiao-Rong Peng, Yanan Wang, Patrick C N Rensen
Cardiovascular Research.2022; 118(2): 489. CrossRef - Obesity-Related Adipose Tissue Remodeling in the Light of Extracellular Mitochondria Transfer
Simon Lecoutre, Karine Clément, Isabelle Dugail
International Journal of Molecular Sciences.2022; 23(2): 632. CrossRef - IL-4 polarized human macrophage exosomes control cardiometabolic inflammation and diabetes in obesity
Tuan Anh Phu, Martin Ng, Ngan K. Vu, Laura Bouchareychas, Robert L. Raffai
Molecular Therapy.2022; 30(6): 2274. CrossRef - Insulin-inducible THRSP maintains mitochondrial function and regulates sphingolipid metabolism in human adipocytes
Maria A. Ahonen, Marcus Höring, Van Dien Nguyen, Sami Qadri, Juuso H. Taskinen, Meghana Nagaraj, Martin Wabitsch, Pamela Fischer-Posovszky, You Zhou, Gerhard Liebisch, P. A. Nidhina Haridas, Hannele Yki-Järvinen, Vesa M. Olkkonen
Molecular Medicine.2022;[Epub] CrossRef - Modulation of adipose inflammation by cellular retinoic acid-binding protein 1
Chin-Wen Wei, Jennifer Nhieu, Yu-Lung Lin, Li-Na Wei
International Journal of Obesity.2022; 46(10): 1759. CrossRef - The Role of Adipokines in Pancreatic Cancer
Qi Wang, Huizhi Wang, Yuntao Ding, Mengtian Wan, Min Xu
Frontiers in Oncology.2022;[Epub] CrossRef - Epigenetic Reprogramming of the Inflammatory Response in Obesity and Type 2 Diabetes
Federica Zatterale, Gregory Alexander Raciti, Immacolata Prevenzano, Alessia Leone, Michele Campitelli, Veronica De Rosa, Francesco Beguinot, Luca Parrillo
Biomolecules.2022; 12(7): 982. CrossRef - Cellular Metabolism and Bioenergetic Function in Human Fibroblasts and Preadipocytes of Type 2 Familial Partial Lipodystrophy
Cristina Algieri, Chiara Bernardini, Fabiana Trombetti, Elisa Schena, Augusta Zannoni, Monica Forni, Salvatore Nesci
International Journal of Molecular Sciences.2022; 23(15): 8659. CrossRef - Shared pathobiology identifies AMPK as a therapeutic target for obesity and autosomal dominant polycystic kidney disease
Ioan-Andrei Iliuta, Xuewen Song, Lauren Pickel, Amirreza Haghighi, Ravi Retnakaran, James Scholey, Hoon-Ki Sung, Gregory R. Steinberg, York Pei
Frontiers in Molecular Biosciences.2022;[Epub] CrossRef - Hypoxia as a Double-Edged Sword to Combat Obesity and Comorbidities
Ruwen Wang, Qin Sun, Xianmin Wu, Yiyin Zhang, Xiaorui Xing, Kaiqing Lin, Yue Feng, Mingqi Wang, Yibing Wang, Ru Wang
Cells.2022; 11(23): 3735. CrossRef - Macrophage and Adipocyte Mitochondrial Dysfunction in Obesity-Induced Metabolic Diseases
Liwen Wang, Jie Hu, Haiyan Zhou
The World Journal of Men's Health.2021; 39(4): 606. CrossRef - ESRRA (estrogen related receptor alpha) is a critical regulator of intestinal homeostasis through activation of autophagic flux via gut microbiota
Sup Kim, June-Young Lee, Seul Gi Shin, Jin Kyung Kim, Prashanta Silwal, Young Jae Kim, Na-Ri Shin, Pil Soo Kim, Minho Won, Sang-Hee Lee, Soo Yeon Kim, Miwa Sasai, Masahiro Yamamoto, Jin-Man Kim, Jin-Woo Bae, Eun-Kyeong Jo
Autophagy.2021; 17(10): 2856. CrossRef - GDF15 as a central mediator for integrated stress response and a promising therapeutic molecule for metabolic disorders and NASH
Kook Hwan Kim, Myung-Shik Lee
Biochimica et Biophysica Acta (BBA) - General Subjects.2021; 1865(3): 129834. CrossRef - The Influence of Obesity and Associated Fatty Acids on Placental Inflammation
Alison J. Eastman, Rebecca E. Moore, Steven D. Townsend, Jennifer A. Gaddy, David M. Aronoff
Clinical Therapeutics.2021; 43(2): 265. CrossRef - Targeting the G protein-coupled estrogen receptor (GPER) in obesity and diabetes
Geetanjali Sharma, Eric R. Prossnitz
Endocrine and Metabolic Science.2021; 2: 100080. CrossRef - Changes in Body Composition Are Associated with Metabolic Changes and the Risk of Metabolic Syndrome
Yun Hwan Oh, Seulggie Choi, Gyeongsil Lee, Joung Sik Son, Kyae Hyung Kim, Sang Min Park
Journal of Clinical Medicine.2021; 10(4): 745. CrossRef - N6-Adenosine Methylation (m6A) RNA Modification: an Emerging Role in Cardiovascular Diseases
Ye-shi Chen, Xin-ping Ouyang, Xiao-hua Yu, Petr Novák, Le Zhou, Ping-ping He, Kai Yin
Journal of Cardiovascular Translational Research.2021; 14(5): 857. CrossRef - From Metabolic Syndrome to Neurological Diseases: Role of Autophagy
Jessica Maiuolo, Micaela Gliozzi, Vincenzo Musolino, Cristina Carresi, Federica Scarano, Saverio Nucera, Miriam Scicchitano, Francesca Bosco, Stefano Ruga, Maria Caterina Zito, Roberta Macri, Rosamaria Bulotta, Carolina Muscoli, Vincenzo Mollace
Frontiers in Cell and Developmental Biology.2021;[Epub] CrossRef - Absent Exercise-Induced Improvements in Fat Oxidation in Women With Polycystic Ovary Syndrome After High-Intensity Interval Training
Sofie Lionett, Ida Almenning Kiel, Ragnhild Røsbjørgen, Stian Lydersen, Steen Larsen, Trine Moholdt
Frontiers in Physiology.2021;[Epub] CrossRef - Roles of interstitial fluid pH and weak organic acids in development and amelioration of insulin resistance
Yoshinori Marunaka
Biochemical Society Transactions.2021; 49(2): 715. CrossRef - The Role of Mitochondrial Adaptation and Metabolic Flexibility in the Pathophysiology of Obesity and Insulin Resistance: an Updated Overview
Dimitrios Tsilingiris, Evangelia Tzeravini, Chrysi Koliaki, Maria Dalamaga, Alexander Kokkinos
Current Obesity Reports.2021; 10(3): 191. CrossRef - Obesity-Related Inflammation and Endothelial Dysfunction in COVID-19: Impact on Disease Severity
Andrea De Lorenzo, Vanessa Estato, Hugo C Castro-Faria-Neto, Eduardo Tibirica
Journal of Inflammation Research.2021; Volume 14: 2267. CrossRef - Thermogenic Fat: Development, Physiological Function, and Therapeutic Potential
Bruna B. Brandão, Ankita Poojari, Atefeh Rabiee
International Journal of Molecular Sciences.2021; 22(11): 5906. CrossRef - Metabolic Syndrome in an Aging Society – Role of Oxidant-Antioxidant Imbalance and Inflammation Markers in Disentangling Atherosclerosis
Sylwia Dziegielewska-Gesiak
Clinical Interventions in Aging.2021; Volume 16: 1057. CrossRef - Recruitment and remodeling of peridroplet mitochondria in human adipose tissue
Rebeca Acín-Perez, Anton Petcherski, Michaela Veliova, Ilan Y. Benador, Essam A. Assali, Georgia Colleluori, Saverio Cinti, Alexandra J. Brownstein, Siyouneh Baghdasarian, Masha J. Livhits, Michael W. Yeh, Karthickeyan Chella Krishnan, Laurent Vergnes, Na
Redox Biology.2021; 46: 102087. CrossRef - New Insights Into Mitochondrial Dysfunction at Disease Susceptibility Loci in the Development of Type 2 Diabetes
Hannah Maude, Winston Lau, Nikolas Maniatis, Toby Andrew
Frontiers in Endocrinology.2021;[Epub] CrossRef - Effects of sleeve gastrectomy on bone mass, microstructure of femurs and bone metabolism associated serum factors in obese rats
Ying Xue, Ran Li, Yong Zhao, Ling Li, Yun Zhou
BMC Endocrine Disorders.2021;[Epub] CrossRef - The cyclin dependent kinase inhibitor Roscovitine prevents diet-induced metabolic disruption in obese mice
Nabil Rabhi, Kathleen Desevin, Briana Noel Cortez, Ryan Hekman, Jean Z. Lin, Andrew Emili, Stephen R. Farmer
Scientific Reports.2021;[Epub] CrossRef - Reliability and variation in mitochondrial respiration in human adipose tissue
Ronni Eg Sahl, Eva Frederikke Høy Helms, Malte Schmücker, Mathias Flensted-Jensen, Arthur Ingersen, Thomas Morville, Flemming Dela, Jørn Wulff Helge, Steen Larsen
Adipocyte.2021; 10(1): 605. CrossRef - Inhibition of protein tyrosine phosphatase improves mitochondrial bioenergetics and dynamics, reduces oxidative stress, and enhances adipogenic differentiation potential in metabolically impaired progenitor stem cells
Katarzyna Kornicka-Garbowska, Lynda Bourebaba, Michael Röcken, Krzysztof Marycz
Cell Communication and Signaling.2021;[Epub] CrossRef - microRNAs in Human Adipose Tissue Physiology and Dysfunction
Alina Kurylowicz
Cells.2021; 10(12): 3342. CrossRef - Aging, obese-insulin resistance, and bone remodeling
Napatsorn Imerb, Chanisa Thonusin, Nipon Chattipakorn, Siriporn C. Chattipakorn
Mechanisms of Ageing and Development.2020; 191: 111335. CrossRef - Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes
Federica Zatterale, Michele Longo, Jamal Naderi, Gregory Alexander Raciti, Antonella Desiderio, Claudia Miele, Francesco Beguinot
Frontiers in Physiology.2020;[Epub] CrossRef - Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases?
Alexis Diaz-Vegas, Pablo Sanchez-Aguilera, James R Krycer, Pablo E Morales, Matías Monsalves-Alvarez, Mariana Cifuentes, Beverly A Rothermel, Sergio Lavandero
Endocrine Reviews.2020;[Epub] CrossRef - Inflammatory Signaling and Brown Fat Activity
Farah Omran, Mark Christian
Frontiers in Endocrinology.2020;[Epub] CrossRef - Omega-3 fatty acids as regulators of brown/beige adipose tissue: from mechanisms to therapeutic potential
Marta Fernández-Galilea, Elisa Félix-Soriano, Ignacio Colón-Mesa, Xavier Escoté, Maria J. Moreno-Aliaga
Journal of Physiology and Biochemistry.2020; 76(2): 251. CrossRef - Anti-Inflammatory Strategies Targeting Metaflammation in Type 2 Diabetes
Alina Kuryłowicz, Krzysztof Koźniewski
Molecules.2020; 25(9): 2224. CrossRef - Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4
Mariana Renovato-Martins, Catharina Moreira-Nunes, Georgia C. Atella, Christina Barja-Fidalgo, João Alfredo de Moraes
Nutrients.2020; 12(9): 2828. CrossRef -
Diabetes and Metabolism Journal in 2020: Good to Great
In-Kyung Jeong
Diabetes & Metabolism Journal.2020; 44(1): 1. CrossRef - The Effect of Silibinin on Protein Expression Profile in White Adipose Tissue of Obese Mice
Fei Wang, Shuchun Chen, Luping Ren, Yichao Wang, Zelin Li, Tiantian Song, He Zhang, Qiwen Yang
Frontiers in Pharmacology.2020;[Epub] CrossRef - Beneficial Effects of Bariatric Surgery-Induced by Weight Loss on the Proteome of Abdominal Subcutaneous Adipose Tissue
Bárbara María Varela-Rodríguez, Paula Juiz-Valiña, Luis Varela, Elena Outeiriño-Blanco, Susana Belén Bravo, María Jesús García-Brao, Enrique Mena, José Francisco Noguera, Javier Valero-Gasalla, Fernando Cordido, Susana Sangiao-Alvarellos
Journal of Clinical Medicine.2020; 9(1): 213. CrossRef - Impact of Skeletal Muscle Mass on Metabolic Health
Gyuri Kim, Jae Hyeon Kim
Endocrinology and Metabolism.2020; 35(1): 1. CrossRef - Sea buckthorn (Hippophae rhamnoides L.) oil enhances proliferation, adipocytes differentiation and insulin sensitivity in 3T3-L1 cells
Ting Zhang, Xuze Qin, Yuxin Cao, Jianxin Zhang, Junxing Zhao
Food Science and Biotechnology.2020; 29(11): 1511. CrossRef - Adipose tissue secretory profile and cardiometabolic risk in obesity
Pengcheng Zhang, Daniels Konja, Yu Wang
Endocrine and Metabolic Science.2020; 1(3-4): 100061. CrossRef - Mitochondrial Dynamics in the Brain Are Associated With Feeding, Glucose Homeostasis, and Whole-Body Metabolism
Jessica L. Haigh, Lauryn E. New, Beatrice M. Filippi
Frontiers in Endocrinology.2020;[Epub] CrossRef - Adipogenesis: A Necessary but Harmful Strategy
Mohammed El Hafidi, Mabel Buelna-Chontal, Fausto Sánchez-Muñoz, Roxana Carbó
International Journal of Molecular Sciences.2019; 20(15): 3657. CrossRef
- Prolonged Endurance Exercise Increases Macrophage Content and Mitochondrial Respiration in Adipose Tissue in Trained Men
- Pathophysiology
- Investigating Susceptibility to Diabetes Using Features of the Adipose Tissue in Response to
In Utero Polycyclic Aromatic Hydrocarbons Exposure - Worlanyo E. Gato, Daniel A. Hunter, Shamaya L. Whitby, Christopher A. Mays, Wilson Yau
- Diabetes Metab J. 2016;40(6):494-508. Published online August 12, 2016
- DOI: https://doi.org/10.4093/dmj.2016.40.6.494
- 3,614 View
- 92 Download
- 7 Web of Science
- 5 Crossref
-
Abstract
PDFSupplementary MaterialPubReader
Background In recent times, there has been an increase in the incidence of type 2 diabetes mellitus (T2DM) particularly in children. Adipocyte dysfunction provide a critical link between obesity and insulin resistance resulting in diabetes outcome. Further, environmental chemical exposure during early years of life might be a significant contributing factor to the increase in the incidence of T2DM. This study tests the idea that exposure to environmental contaminants (2-aminoanthracene [2AA])
in utero will show effects in the adipose tissue (AT) that signify T2DM vulnerability. 2AA is a polycyclic aromatic hydrocarbon found in a variety of products.Methods To accomplish the study objective, pregnant dams were fed various amounts of 2AA adulterated diets from gestation through postnatal period. The neonates and older offspring were analyzed for diabetic-like genes in the ATs and analysis of serum glucose. Furthermore, weight monitoring, histopathology and immunohistochemical (IHC) staining for CD68 in AT, adipocyte size determination and adiponectin amounts in serum were undertaken.
Results Up-regulation of adiponectin and interleukin-6 genes were noted in the pups and older rats. Combination of intrauterine 2AA toxicity with moderate high fat diet exhibited gene expression patterns similar to those of the neonates. Elevated serum glucose levels were noted in treated groups. IHC of the AT indicated no significant malformations; however, CD68+ cells were greater in the animals treated to 2AA. Similarly, mean sizes of the adipocytes were larger in treated and combined 2AA and moderate high fat animals. Adiponectin was reduced in 2AA groups.
Conclusion From the preceding, it appears intrauterine 2AA disturbance, when combined with excess fat accumulation will lead to greater risk for the diabetic condition.
-
Citations
Citations to this article as recorded by- The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies
Jan Aaseth, Dragana Javorac, Aleksandra Djordjevic, Zorica Bulat, Anatoly Skalny, Irina Zaitseva, Michael Aschner, Alexey Tinkov
Toxics.2022; 10(2): 65. CrossRef - The Association between Urinary Polycyclic Aromatic Hydrocarbons Metabolites and Type 2 Diabetes Mellitus
Xue Wang, Ang Li, Qun Xu
International Journal of Environmental Research and Public Health.2022; 19(13): 7605. CrossRef - Dietary ingestion of 2-aminoanthracene (2AA) and the risk for type-1 diabetes (T1D)
Isaiah Seise, Zachary A. Pilz, Moses Yeboah Kusi, Bethany Bogan, Brittany Jean McHale, Worlanyo E. Gato
Journal of Environmental Science and Health, Part A.2020; 55(14): 1638. CrossRef - Association of the IL6 Gene Polymorphism with Component Features of Metabolic Syndrome in Obese Subjects
Elham Barati, Hamideh Ghazizadeh, Fatemeh Sadabadi, Elham Kazemi, Gordon A. Ferns, Amir Avan, Majid Ghayour-Mobarhan
Biochemical Genetics.2019; 57(5): 695. CrossRef - The hepatic effects in dams that ingested 2-aminoanthracene during gestation and lactation
Raven E Ulieme, Surjania Awer, John C Stagg, Wilson Yau, Worlanyo E Gato
Toxicology and Industrial Health.2019; 35(9): 568. CrossRef
- The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies
- Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue
- Chang Yeop Han
- Diabetes Metab J. 2016;40(4):272-279. Published online June 28, 2016
- DOI: https://doi.org/10.4093/dmj.2016.40.4.272
- 5,311 View
- 91 Download
- 112 Web of Science
- 112 Crossref
-
Abstract
PDFPubReader
Obesity resulting from the delivery of an excess amount of energy to adipose tissue from glucose or free fatty acids is associated with insulin resistance and adipose tissue inflammation. Reactive oxygen species (ROS) have been implicated as contributors to both the onset and the progression of insulin resistance. ROS can be generated by overloading the mitochondrial oxidative phosphorylation system, and also by nicotinamide adenine dinucleotide phosphate oxidases (NOX) produced by either adipocytes, which only produce NOX4, or by macrophages, which produce mainly NOX2. The source of the ROS might differ in the early, intermediate and late stages of obesity, switching from NOX4-dependence in the early phases to NOX2-dependence, in the intermediate phase, and transiting to mitochondria-dependence later in the time course of obesity. Thus, depending on the stage of obesity, ROS can be generated by three distinct mechanisms: i.e., NOX4, NOX2, and mitochondria. In this review, we will discuss whether NOX4-, NOX2-, and/or mitochondria-derived ROS is/are causal in the onset of adipocyte insulin resistance as obesity progresses. Moreover, we will review the pathophysiological roles of NOX4, NOX2, and mitochondria-derived ROS on adipose tissue inflammation.
-
Citations
Citations to this article as recorded by- The Relationship of Alcohol Consumption and Drinking Pattern to the Risk of Glomerular Hyperfiltration in Middle-aged Japanese Men: The Kansai Healthcare Study
Mikiko Shibata, Kyoko Kogawa Sato, Hideo Koh, Izumi Shibata, Kaori Okamura, Yuka Takeuchi, Keiko Oue, Michio Morimoto, Tomoshige Hayashi
Journal of Epidemiology.2024; 34(3): 137. CrossRef - Monoamine oxidase mediated oxidative stress: a potential molecular and biochemical crux in the pathogenesis of obesity
J. P. Shirley Niveta, Cordelia Mano John, Sumathy Arockiasamy
Molecular Biology Reports.2024;[Epub] CrossRef - The obesity-autophagy-cancer axis: Mechanistic insights and therapeutic perspectives
Amir Barzegar Behrooz, Marco Cordani, Alessandra Fiore, Massimo Donadelli, Joseph W. Gordon, Daniel J. Klionsky, Saeid Ghavami
Seminars in Cancer Biology.2024; 99: 24. CrossRef - Pathogenesis and management of diabetic gastroparesis: An updated clinically oriented review
Srikar Uppaluri, Manisha Ashok Jain, Hira Ali, Jay Shingala, Dhruti Amin, Trisha Ajwani, Irum Fatima, Neil Patel, Nirja Kaka, Yashendra Sethi, Nitin Kapoor
Diabetes & Metabolic Syndrome: Clinical Research & Reviews.2024; 18(3): 102994. CrossRef - The advent of RNA-based therapeutics for metabolic syndrome and associated conditions: a comprehensive review of the literature
Helen Ye Rim Huang, Sarah Badar, Mohammad Said, Siddiqah Shah, Hareesha Rishab Bharadwaj, Krishna Ramamoorthy, Maen Monketh Alrawashdeh, Faaraea Haroon, Jawad Basit, Sajeel Saeed, Narjiss Aji, Gary Tse, Priyanka Roy, Mainak Bardhan
Molecular Biology Reports.2024;[Epub] CrossRef - The complex interplay between oxinflammation, mitochondrial dysfunction and lipotoxicity: Focus on their role in the pathogenesis of skeletal muscle insulin resistance and modulation by dietary fatty acids
Angelina Passaro, Juana Maria Sanz, Nenad Naumovski, Domenico Sergi
Advances in Redox Research.2024; 11: 100100. CrossRef - Extracellular vesicles regulate the transmission of insulin resistance and redefine noncommunicable diseases
Biao Li, Wan Li, Tiancai Liu, Longying Zha
Frontiers in Molecular Biosciences.2023;[Epub] CrossRef - Oxidative stress in metabolic diseases: current scenario and therapeutic relevance
Satish K. Raut, Madhu Khullar
Molecular and Cellular Biochemistry.2023; 478(1): 185. CrossRef - Rainbow Trout (Oncorhynchus mykiss) as Source of Multifunctional Peptides with Antioxidant, ACE and DPP-IV Inhibitory Activities
Martina Bartolomei, Janna Cropotova, Carlotta Bollati, Kristine Kvangarsnes, Lorenza d’Adduzio, Jianqiang Li, Giovanna Boschin, Carmen Lammi
Nutrients.2023; 15(4): 829. CrossRef - Current Status of Obesity: Protective Role of Catechins
Tanisha Basu, Ashley Selman, Arubala P. Reddy, P. Hemachandra Reddy
Antioxidants.2023; 12(2): 474. CrossRef - Advanced Oxidation Protein Products Contribute to Chronic-Kidney-Disease-Induced Adipose Inflammation through Macrophage Activation
Nanaka Arimura, Hiroshi Watanabe, Hiromasa Kato, Tadashi Imafuku, Takehiro Nakano, Miyu Sueyoshi, Mayuko Chikamatsu, Kai Tokumaru, Taisei Nagasaki, Hitoshi Maeda, Motoko Tanaka, Kazutaka Matsushita, Toru Maruyama
Toxins.2023; 15(3): 179. CrossRef - The Role of Oxidative Stress Enhanced by Adiposity in Cardiometabolic Diseases
Iwona Świątkiewicz, Marcin Wróblewski, Jarosław Nuszkiewicz, Paweł Sutkowy, Joanna Wróblewska, Alina Woźniak
International Journal of Molecular Sciences.2023; 24(7): 6382. CrossRef - Understanding the Impact of Obesity on Ageing in the Radiance of DNA Metabolism
S.G. Chowdhury, S. Misra, Parimal Karmakar
The Journal of nutrition, health and aging.2023; 27(5): 314. CrossRef - Cross-talk between insulin resistance and nitrogen species in hypoxia leads to deterioration of tissue and homeostasis
Priyanshy Sharma, V. Sri Swetha Victoria, P. Praneeth Kumar, Sarbani Karmakar, Mudduluru Swetha, Amala Reddy
International Immunopharmacology.2023; 122: 110472. CrossRef - Mechanisms of Formation and Persistence of IgE Products and Potential Innovative Means of Therapy for Allergic Pathologies
D. B. Chudakov, M. V. Konovalova, M. A. Streltsova, O. A. Shustova, A. A. Generalov, G. V. Fattakhova
Applied Biochemistry and Microbiology.2023; 59(6): 754. CrossRef - Mechanism and application of Lactobacillus in type 2 diabetes-associated periodontitis
Sisi Chen, Yuhan Zhang
Frontiers in Public Health.2023;[Epub] CrossRef - Activated monocytes as a therapeutic target to attenuate vascular inflammation and lower cardiovascular disease-risk in patients with type 2 diabetes: A systematic review of preclinical and clinical studies
Siphamandla R. Ngcobo, Bongani B. Nkambule, Tawanda M. Nyambuya, Kabelo Mokgalaboni, Aviwe Ntsethe, Vuyolwethu Mxinwa, Khanyisani Ziqubu, Yonela Ntamo, Thembeka A. Nyawo, Phiwayinkosi V. Dludla
Biomedicine & Pharmacotherapy.2022; 146: 112579. CrossRef - Genomic Analysis of Visceral Fat Accumulation in Holstein Cows
Larissa C. Novo, Ligia Cavani, Pablo Pinedo, Pedro Melendez, Francisco Peñagaricano
Frontiers in Genetics.2022;[Epub] CrossRef - Regulation of immune cell function by nicotinamide nucleotide transhydrogenase
Thomas Regan, Rachel Conway, Leena P. Bharath
American Journal of Physiology-Cell Physiology.2022; 322(4): C666. CrossRef - d-Allulose Ameliorates Hyperglycemia Through IRE1α Sulfonation-RIDD-Sirt1 Decay Axis in the Skeletal Muscle
Hwa-Young Lee, Geum-Hwa Lee, The-Hiep Hoang, Seon-Ah Park, Juwon Lee, Junghyun Lim, Soonok Sa, Go Eun Kim, Jung Sook Han, Junghyun Kim, Han-Jung Chae
Antioxidants & Redox Signaling.2022; 37(4-6): 229. CrossRef - Impact of physical exercise and caloric restriction in patients with type 2 diabetes: Skeletal muscle insulin resistance and mitochondrial dysfunction as ideal therapeutic targets
Sinenhlanhla X.H. Mthembu, Sithandiwe E. Mazibuko-Mbeje, Khanyisani Ziqubu, Thembeka A. Nyawo, Nnini Obonye, Tawanda M. Nyambuya, Bongani B. Nkambule, Sonia Silvestri, Luca Tiano, Christo J.F. Muller, Phiwayinkosi V. Dludla
Life Sciences.2022; 297: 120467. CrossRef - The burden of overweight: Higher body mass index, but not vital exhaustion, is associated with higher DNA damage and lower DNA repair capacity
Judy Fieres, Marvin Fischer, Christine Sauter, Maria Moreno-Villanueva, Alexander Bürkle, Petra H. Wirtz
DNA Repair.2022; 114: 103323. CrossRef - The Disordered Amino Terminus of the Circadian Enzyme Nocturnin Modulates Its NADP(H) Phosphatase Activity by Changing Protein Dynamics
Anushka C. Wickramaratne, Li Li, Jesse B. Hopkins, Lukasz A. Joachimiak, Carla B. Green
Biochemistry.2022; 61(11): 1091. CrossRef - Oxidative stress and obesity
Maja Malenica, Neven Meseldžić
Arhiv za farmaciju.2022; 72(2): 166. CrossRef - Transcriptomic analysis of Simpson Golabi Behmel Syndrome cells during differentiation exhibit BAT-like function
M. Colitti, U. Ali, M. Wabitsch, D. Tews
Tissue and Cell.2022; : 101822. CrossRef - Vitamin E supplementation improves testosterone, glucose- and lipid-related metabolism in women with polycystic ovary syndrome: a meta-analysis of randomized clinical trials
Sebastián Yalle-Vásquez, Karem Osco-Rosales, Wendy Nieto-Gutierrez, Vicente Benites-Zapata, Faustino R. Pérez-López, Christoper A. Alarcon-Ruiz
Gynecological Endocrinology.2022; 38(7): 548. CrossRef - The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity
Orestes López-Ortega, Nidia Carolina Moreno-Corona, Victor Javier Cruz-Holguin, Luis Didier Garcia-Gonzalez, Addy Cecilia Helguera-Repetto, Mirza Romero-Valdovinos, Haruki Arevalo-Romero, Leticia Cedillo-Barron, Moisés León-Juárez
International Journal of Molecular Sciences.2022; 23(11): 6154. CrossRef - Participation of Magnesium in the Secretion and Signaling Pathways of Insulin: an Updated Review
Stéfany Rodrigues de Sousa Melo, Loanne Rocha dos Santos, Tamires da Cunha Soares, Bruna Emanuele Pereira Cardoso, Thaline Milany da Silva Dias, Jennifer Beatriz Silva Morais, Mickael de Paiva Sousa, Thayanne Gabryelle Visgueira de Sousa, Nilmara Cunha da
Biological Trace Element Research.2022; 200(8): 3545. CrossRef - Nrf2a dependent and independent effects of early life exposure to 3,3’-dichlorobiphenyl (PCB-11) in zebrafish (Danio rerio)
Monika A. Roy, Charlotte K. Gridley, Sida Li, Yeonhwa Park, Alicia R. Timme-Laragy
Aquatic Toxicology.2022; 249: 106219. CrossRef - Highly active antiretroviral therapy-silver nanoparticle conjugate interacts with neuronal and glial cells and alleviates anxiety-like behaviour in streptozotocin-induced diabetic rats
Sodiq Kolawole Lawal, Samuel Oluwaseun Olojede, Ayobami Dare, Oluwaseun Samuel Faborode, Sheu Oluwadare Sulaiman, Edwin Coleridge Naidu, Carmen Olivia Rennie, Onyemaechi Okpara Azu
IBRO Neuroscience Reports.2022; 13: 57. CrossRef - Vitamin C attenuates predisposition to high-fat diet-induced metabolic dysregulation in GLUT10-deficient mouse model
Chung-Lin Jiang, Chang-Yu Tsao, Yi-Ching Lee
Genes & Nutrition.2022;[Epub] CrossRef - Adipose Tissue Aging and Metabolic Disorder, and the Impact of Nutritional Interventions
Xiujuan Wang, Meihong Xu, Yong Li
Nutrients.2022; 14(15): 3134. CrossRef - Production of Reactive Oxygen Species by Epicardial Adipocytes Is Associated with an Increase in Postprandial Glycemia, Postprandial Insulin, and a Decrease in Serum Adiponectin in Patients with Severe Coronary Atherosclerosis
Natalia V. Naryzhnaya, Olga A. Koshelskaya, Irina V. Kologrivova, Tatiana E. Suslova, Olga A. Kharitonova, Sergey L. Andreev, Alexander S. Gorbunov, Boris K. Kurbatov, Alla A. Boshchenko
Biomedicines.2022; 10(8): 2054. CrossRef - Aberrant mitochondrial homeostasis at the crossroad of musculoskeletal ageing and non-small cell lung cancer
Konstantinos Prokopidis, Panagiotis Giannos, Oliver C. Witard, Daniel Peckham, Theocharis Ispoglou, Ajay Pratap Singh
PLOS ONE.2022; 17(9): e0273766. CrossRef - Nutraceutical Prevention of Diabetic Complications—Focus on Dicarbonyl and Oxidative Stress
Mark F. McCarty, James J. DiNicolantonio, James H. O’Keefe
Current Issues in Molecular Biology.2022; 44(9): 4314. CrossRef - Fructose promotes more than glucose the adipocytic differentiation of pig mesenchymal stem cells
Francisco Campos‐Maldonado, María L. González‐Dávalos, Enrique Piña, Miriam Aracely Anaya‐Loyola, Armando Shimada, Alfredo Varela‐Echavarria, Ofelia Mora
Journal of Food Biochemistry.2022;[Epub] CrossRef - Understanding the effect of obesity on papillary thyroid cancer: is there a need for tailored diagnostic and therapeutic management?
Antonio Matrone, Alessio Basolo, Ferruccio Santini, Rossella Elisei
Expert Review of Endocrinology & Metabolism.2022; 17(6): 475. CrossRef - The Role of Inflammation as a Preponderant Risk Factor in Cardiovascular
Diseases
Rodrigo Damián García, Joana Antonela Asensio, Diahann Jeanette Perdicaro, María de los Ángeles Peral
Current Vascular Pharmacology.2022; 20(3): 244. CrossRef - Synthesis and Catalytic Studies of Nanoalloy Particles Based on Bismuth, Silver, and Rhenium
Konrad Wojtaszek, Katarzyna Skibińska, Filip Cebula, Tomasz Tokarski, Marc Escribà-Gelonch, Volker Hessel, Marek Wojnicki
Metals.2022; 12(11): 1819. CrossRef - Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review
Francesco Nappi, Antonio Fiore, Joyce Masiglat, Teresa Cavuoti, Michela Romandini, Pierluigi Nappi, Sanjeet Singh Avtaar Singh, Jean-Paul Couetil
Biomedicines.2022; 10(11): 2884. CrossRef - MicroRNAs in the Regulation of NADPH Oxidases in Vascular Diabetic and Ischemic Pathologies: A Case for Alternate Inhibitory Strategies?
Sean R. Wallace, Patrick J. Pagano, Damir Kračun
Antioxidants.2022; 12(1): 70. CrossRef - Potential Roles of Adipocyte Extracellular Vesicle–Derived miRNAs in Obesity-Mediated Insulin Resistance
Yujeong Kim, Ok-Kyung Kim
Advances in Nutrition.2021; 12(2): 566. CrossRef - Mesenchymal stem cell conditioned medium ameliorates diabetic serum‐induced insulin resistance in 3T3‐L1 cells
Avinash Sanap, Ramesh Bhonde, Kalpana Joshi
Chronic Diseases and Translational Medicine.2021; 7(1): 47. CrossRef - F13A1 transglutaminase expression in human adipose tissue increases in acquired excess weight and associates with inflammatory status of adipocytes
M. T. Kaartinen, M. Arora, S. Heinonen, A. Hang, A. Barry, J. Lundbom, A. Hakkarainen, N. Lundholm, A. Rissanen, J. Kaprio, K. H. Pietiläinen
International Journal of Obesity.2021; 45(3): 577. CrossRef - Obesity and the prevention of thyroid cancer: Impact of body mass index and weight change on developing thyroid cancer – Pooled results of 24 million cohorts
Mohanad R. Youssef, Adin S.C. Reisner, Abdallah S. Attia, Mohamed Hosny Hussein, Mahmoud Omar, Anna LaRussa, Carlos A. Galvani, Mohamed Aboueisha, Mohamed Abdelgawad, Eman Ali Toraih, Gregory W. Randolph, Emad Kandil
Oral Oncology.2021; 112: 105085. CrossRef - Thioredoxin deficiency exacerbates vascular dysfunction during diet‐induced obesity in small mesenteric artery in mice
Shannon Dunn, Robert H. Hilgers, Kumuda C. Das
Microcirculation.2021;[Epub] CrossRef - Protein Digests and Pure Peptides from Chia Seed Prevented Adipogenesis and Inflammation by Inhibiting PPARγ and NF-κB Pathways in 3T3L-1 Adipocytes
Mariana Grancieri, Hércia Stampini Duarte Martino, Elvira Gonzalez de Mejia
Nutrients.2021; 13(1): 176. CrossRef - The Interplay Between Adipose Tissue and Vasculature: Role of Oxidative Stress in Obesity
Yawen Zhou, Huige Li, Ning Xia
Frontiers in Cardiovascular Medicine.2021;[Epub] CrossRef - Peri-renal adipose inflammation contributes to renal dysfunction in a non-obese prediabetic rat model: Role of anti-diabetic drugs
Safaa H. Hammoud, Ibrahim AlZaim, Nahed Mougharbil, Sahar Koubar, Ali H. Eid, Assaad A. Eid, Ahmed F. El-Yazbi
Biochemical Pharmacology.2021; 186: 114491. CrossRef - Hydrogen Nano-Bubble Water Suppresses ROS Generation, Adipogenesis, and Interleukin-6 Secretion in Hydrogen-Peroxide- or PMA-Stimulated Adipocytes and Three-Dimensional Subcutaneous Adipose Equivalents
Li Xiao, Nobuhiko Miwa
Cells.2021; 10(3): 626. CrossRef - Redox Homeostasis in Pancreatic β-Cells: From Development to Failure
Štěpánka Benáková, Blanka Holendová, Lydie Plecitá-Hlavatá
Antioxidants.2021; 10(4): 526. CrossRef - Htd2 deficiency-associated suppression of α-lipoic acid production provokes mitochondrial dysfunction and insulin resistance in adipocytes
Mengqi Zeng, Jie Xu, Zhengyi Zhang, Xuan Zou, Xueqiang Wang, Ke Cao, Weiqiang Lv, Yuting Cui, Jiangang Long, Zhihui Feng, Jiankang Liu
Redox Biology.2021; 41: 101948. CrossRef - Nutraceutical, Dietary, and Lifestyle Options for Prevention and Treatment of Ventricular Hypertrophy and Heart Failure
Mark F. McCarty
International Journal of Molecular Sciences.2021; 22(7): 3321. CrossRef - Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry
Pamela A. Nono Nankam, Télesphore B. Nguelefack, Julia H. Goedecke, Matthias Blüher
Antioxidants.2021; 10(4): 622. CrossRef - Effects of different models of sucrose intake on the oxidative status of the uterus and ovary of rats
Joanna Sadowska, Wioleta Dudzińska, Izabela Dziaduch, Mahmoud A.O. Dawood
PLOS ONE.2021; 16(5): e0251789. CrossRef - Oxidative stress in oocyte aging and female reproduction
Ling Wang, Jinhua Tang, Lei Wang, Feng Tan, Huibin Song, Jiawei Zhou, Fenge Li
Journal of Cellular Physiology.2021; 236(12): 7966. CrossRef - Temporal correlation of morphological and biochemical changes with the recruitment of different mechanisms of reactive oxygen species formation during human SW872 cell adipogenic differentiation
Mara Fiorani, Rita De Matteis, Barbara Canonico, Giulia Blandino, Alessandro Mazzoli, Mariele Montanari, Andrea Guidarelli, Orazio Cantoni
BioFactors.2021; 47(5): 837. CrossRef - Antioxidant and anti-inflammatory properties of gamma- oryzanol attenuates insulin resistance by increasing GLUT- 4 expression in skeletal muscle of obese animals
Letícia Mattei, Fabiane Valentini Francisqueti-Ferron, Jéssica Leite Garcia, Artur Junio Togneri Ferron, Carol Cristina Vágula de Almeida Silva, Cristina Schmitt Gregolin, Erika Tiemi Nakandakare-Maia, Janaína das Chagas Paixão Silva, Fernando Moreto, Igo
Molecular and Cellular Endocrinology.2021; 537: 111423. CrossRef - Serum Bilirubin Levels in Overweight and Obese Individuals: The Importance of Anti-Inflammatory and Antioxidant Responses
Lovro Žiberna, Zala Jenko-Pražnikar, Ana Petelin
Antioxidants.2021; 10(9): 1352. CrossRef - The level of reactive oxygen species production by adipocytes of epicardial adipose tissue is associated with an increase in postprandial glycemia in patients with severe coronary atherosclerosis
O. A. Koshelskaya, N. V. Naryzhnaya, I. V. Kologrivova, T. E. Suslova, O. A. Kharitonova, V. V. Evtushenko, S. L. Andreev, A. S. Gorbunov, A. A. Gudkova
The Siberian Journal of Clinical and Experimental Medicine.2021; 36(3): 59. CrossRef - Exercise training results in depot-specific adaptations to adipose tissue mitochondrial function
Amy E. Mendham, Steen Larsen, Cindy George, Kevin Adams, Jon Hauksson, Tommy Olsson, Melony C. Fortuin-de Smidt, Pamela A. Nono Nankam, Olah Hakim, Louise M. Goff, Carmen Pheiffer, Julia H. Goedecke
Scientific Reports.2020;[Epub] CrossRef - Oxidative stress resulting from the removal of endogenous catalase induces obesity by promoting hyperplasia and hypertrophy of white adipocytes
Su-Kyung Shin, Hyun-Woo Cho, Seung-Eun Song, Seung-Soon Im, Jae-Hoon Bae, Dae-Kyu Song
Redox Biology.2020; 37: 101749. CrossRef - Pathogenic Role of Air Pollution Particulate Matter in Cardiometabolic Disease: Evidence from Mice and Humans
Timoteo Marchini, Andreas Zirlik, Dennis Wolf
Antioxidants & Redox Signaling.2020; 33(4): 263. CrossRef - Fumarate and oxidative stress synergize to promote stability of C/EBP homologous protein in the adipocyte
Allison M. Manuel, Michael D. Walla, Margaret T. Dorn, Ross M. Tanis, Gerardo G. Piroli, Norma Frizzell
Free Radical Biology and Medicine.2020; 148: 70. CrossRef - TNF-α G-308A genetic variants, serum CRP-hs concentration and DNA damage in obese women
Marta Włodarczyk, Michał Ciebiera, Grażyna Nowicka
Molecular Biology Reports.2020; 47(2): 855. CrossRef - Lipolysis modulates the biosynthesis of inflammatory lipid mediators derived from linoleic acid in adipose tissue of periparturient dairy cows
G. Andres Contreras, Jenne De Koster, Jonas de Souza, Juliana Laguna, Vengai Mavangira, Rahul K. Nelli, Jeff Gandy, Adam L. Lock, Lorraine M. Sordillo
Journal of Dairy Science.2020; 103(2): 1944. CrossRef - Dietary Fat and Cancer—Which Is Good, Which Is Bad, and the Body of Evidence
Bianka Bojková, Pawel J. Winklewski, Magdalena Wszedybyl-Winklewska
International Journal of Molecular Sciences.2020; 21(11): 4114. CrossRef - Adipose tissue macrophages: Unique polarization and bioenergetics in obesity
Heather L. Caslin, Monica Bhanot, W. Reid Bolus, Alyssa H. Hasty
Immunological Reviews.2020; 295(1): 101. CrossRef - Conditioned medium of adipose derived Mesenchymal Stem Cells reverse insulin resistance through downregulation of stress induced serine kinases
Avinash Sanap, Ramesh Bhonde, Kalpana Joshi
European Journal of Pharmacology.2020; 881: 173215. CrossRef - Quercetin metabolites from Hibiscus sabdariffa contribute to alleviate glucolipotoxicity-induced metabolic stress in vitro
María Herranz-López, Mariló Olivares-Vicente, Esther Rodríguez Gallego, Jose Antonio Encinar, Almudena Pérez-Sánchez, Verónica Ruiz-Torres, Jorge Joven, Enrique Roche, Vicente Micol
Food and Chemical Toxicology.2020; 144: 111606. CrossRef - Elastin-derived peptide VGVAPG decreases differentiation of mouse embryo fibroblast (3T3-L1) cells into adipocytes
Konrad A. Szychowski, Bartosz Skóra, Jakub Tobiasz, Jan Gmiński
Adipocyte.2020; 9(1): 234. CrossRef - Pharmacological potential of the combination of Salvia miltiorrhiza (Danshen) and Carthamus tinctorius (Honghua) for diabetes mellitus and its cardiovascular complications
John O. Orgah, Shuang He, Yule Wang, Miaomiao Jiang, Yuefei Wang, Emmanuel A. Orgah, Yajun Duan, Buchang Zhao, Boli Zhang, Jihong Han, Yan Zhu
Pharmacological Research.2020; 153: 104654. CrossRef - Microbiota-Mitochondria Inter-Talk: A Potential Therapeutic Strategy in Obesity and Type 2 Diabetes
Teresa Vezza, Zaida Abad-Jiménez, Miguel Marti-Cabrera, Milagros Rocha, Víctor Manuel Víctor
Antioxidants.2020; 9(9): 848. CrossRef - Transglutaminases and Obesity in Humans: Association of F13A1 to Adipocyte Hypertrophy and Adipose Tissue Immune Response
Mari T. Kaartinen, Mansi Arora, Sini Heinonen, Aila Rissanen, Jaakko Kaprio, Kirsi H. Pietiläinen
International Journal of Molecular Sciences.2020; 21(21): 8289. CrossRef - Phytosterols: Nutritional Health Players in the Management of Obesity and Its Related Disorders
Teresa Vezza, Francisco Canet, Aranzazu M. de Marañón, Celia Bañuls, Milagros Rocha, Víctor Manuel Víctor
Antioxidants.2020; 9(12): 1266. CrossRef - Metabolic programming of macrophage functions and pathogens control
Sue-jie Koo, Nisha J. Garg
Redox Biology.2019; 24: 101198. CrossRef - Effects of bariatric surgery on telomere length and T-cell aging
F. Jongbloed, R. W. J. Meijers, J. N. M. IJzermans, R. A. Klaassen, M. E. T. Dollé, S. van den Berg, M. G. H. Betjes, R. W. F. de Bruin, E. van der Harst, N. H. R. Litjens
International Journal of Obesity.2019; 43(11): 2189. CrossRef - Shared pathways for neuroprogression and somatoprogression in neuropsychiatric disorders
Gerwyn Morris, Basant K. Puri, Adam J. Walker, Michael Maes, Andre F. Carvalho, Chiara C. Bortolasci, Ken Walder, Michael Berk
Neuroscience & Biobehavioral Reviews.2019; 107: 862. CrossRef - Adipose oxidative stress and protein carbonylation
Amy K. Hauck, Yimao Huang, Ann V. Hertzel, David A. Bernlohr
Journal of Biological Chemistry.2019; 294(4): 1083. CrossRef - Modulatory functions of bioactive fruits, vegetables and spices in adipogenesis and angiogenesis
Priyanka Sarkar, Kavitha Thirumurugan
Journal of Functional Foods.2019; 53: 318. CrossRef - Spontaneous ketonuria and risk of incident diabetes: a 12 year prospective study
Gyuri Kim, Sang-Guk Lee, Byung-Wan Lee, Eun Seok Kang, Bong-Soo Cha, Ele Ferrannini, Yong-ho Lee, Nam H. Cho
Diabetologia.2019; 62(5): 779. CrossRef - Obesity, DNA Damage, and Development of Obesity-Related Diseases
Marta Włodarczyk, Grażyna Nowicka
International Journal of Molecular Sciences.2019; 20(5): 1146. CrossRef - Weight change is significantly associated with risk of thyroid cancer: A nationwide population-based cohort study
Hyemi Kwon, Kyung-Do Han, Cheol-Young Park
Scientific Reports.2019;[Epub] CrossRef - The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology
Evan DeVallance, Yao Li, Michael J. Jurczak, Eugenia Cifuentes-Pagano, Patrick J. Pagano
Antioxidants & Redox Signaling.2019; 31(10): 687. CrossRef - Phenolic compounds as natural and multifunctional anti-obesity agents: A review
Celia Rodríguez-Pérez, Antonio Segura-Carretero, María del Mar Contreras
Critical Reviews in Food Science and Nutrition.2019; 59(8): 1212. CrossRef - Oxygenic metabolism in nutritional obesity induced by olive oil. The influence of vitamin C
Eraci Drehmer, Mari Ángeles Navarro-Moreno, Sandra Carrera, Vincent M. Villar, Mari Luz Moreno
Food & Function.2019; 10(6): 3567. CrossRef - High Glucose Level Impairs Human Mature Bone Marrow Adipocyte Function Through Increased ROS Production
Tareck Rharass, Stéphanie Lucas
Frontiers in Endocrinology.2019;[Epub] CrossRef - Redox TRPs in diabetes and diabetic complications: Mechanisms and pharmacological modulation
Pratik Adhya, Shyam Sunder Sharma
Pharmacological Research.2019; 146: 104271. CrossRef - Linking Arrhythmias and Adipocytes: Insights, Mechanisms, and Future Directions
Maria A. Pabon, Kevin Manocha, Jim W. Cheung, James C. Lo
Frontiers in Physiology.2018;[Epub] CrossRef - Strawberry extract attenuates oxidative stress in 3T3-L1 cells
Tamara Y. Forbes-Hernández, Sadia Afrin, Danila Cianciosi, Piera Pia Manna, Jiaojiao Zhang, Massimiliano Gasparrini, Patricia Reboredo-Rodríguez
Journal of Berry Research.2018; 8(3): 193. CrossRef - The mitochondrial unfolded protein response and mitohormesis: a perspective on metabolic diseases
Hyon-Seung Yi, Joon Young Chang, Minho Shong
Journal of Molecular Endocrinology.2018; 61(3): R91. CrossRef - Emerging Roles for Adipose Tissue in Cardiovascular Disease
Elizabeth E. Ha, Robert C. Bauer
Arteriosclerosis, Thrombosis, and Vascular Biology.2018;[Epub] CrossRef - miR-199a-3p regulates brown adipocyte differentiation through mTOR signaling pathway
Yao Gao, Yan Cao, Xianwei Cui, Xingyun Wang, Yahui Zhou, Fangyan Huang, Xing Wang, Juan Wen, Kaipeng Xie, Pengfei Xu, Xirong Guo, Lianghui You, Chenbo Ji
Molecular and Cellular Endocrinology.2018; 476: 155. CrossRef - Reduced expression of Twist 1 is protective against insulin resistance of adipocytes and involves mitochondrial dysfunction
Sumei Lu, Hong Wang, Rui Ren, Xiaohong Shi, Yanmei Zhang, Wanshan Ma
Scientific Reports.2018;[Epub] CrossRef - Zanthoxylum ailanthoides Suppresses Oleic Acid-Induced Lipid Accumulation through an Activation of LKB1/AMPK Pathway in HepG2 Cells
Eun-Bin Kwon, Myung-Ji Kang, Soo-Yeon Kim, Yong-Moon Lee, Mi-Kyeong Lee, Heung Joo Yuk, Hyung Won Ryu, Su Ui Lee, Sei-Ryang Oh, Dong-Oh Moon, Hyun-Sun Lee, Mun-Ock Kim
Evidence-Based Complementary and Alternative Medicine.2018; 2018: 1. CrossRef - Supplementation of Lactobacillus plantarum Improves Markers of Metabolic Dysfunction Induced by a High Fat Diet
Alice Martinic, Javad Barouei, Zach Bendiks, Darya Mishchuk, Dustin D. Heeney, Roy Martin, Maria L. Marco, Carolyn M. Slupsky
Journal of Proteome Research.2018; 17(8): 2790. CrossRef - The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro
Xuan Wang, Andris Elksnis, Per Wikström, Erik Walum, Nils Welsh, Per-Ola Carlsson, Harald H. H. W. Schmidt
PLOS ONE.2018; 13(9): e0204271. CrossRef - Involvement of adipose tissue inflammation and dysfunction in virus-induced type 1 diabetes
James C Needell, Madalyn N Brown, Danny Zipris
Journal of Endocrinology.2018; 238(1): 61. CrossRef - Autophagy as an emerging target in cardiorenal metabolic disease: From pathophysiology to management
Yingmei Zhang, Adam T. Whaley-Connell, James R. Sowers, Jun Ren
Pharmacology & Therapeutics.2018; 191: 1. CrossRef - Inflammation and Oxidative Stress in an Obese State and the Protective Effects of Gallic Acid
Phiwayinkosi Dludla, Bongani Nkambule, Babalwa Jack, Zibusiso Mkandla, Tinashe Mutize, Sonia Silvestri, Patrick Orlando, Luca Tiano, Johan Louw, Sithandiwe Mazibuko-Mbeje
Nutrients.2018; 11(1): 23. CrossRef - Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity
Laura J. Den Hartigh, Mohamed Omer, Leela Goodspeed, Shari Wang, Tomasz Wietecha, Kevin D. O’Brien, Chang Yeop Han
Arteriosclerosis, Thrombosis, and Vascular Biology.2017; 37(3): 466. CrossRef - Novel lipid-mimetic prodrugs delivering active compounds to adipose tissue
Andrea Mattarei, Andrea Rossa, Veronica Bombardelli, Michele Azzolini, Martina La Spina, Cristina Paradisi, Mario Zoratti, Lucia Biasutto
European Journal of Medicinal Chemistry.2017; 135: 77. CrossRef - Combined metformin and insulin treatment reverses metabolically impaired omental adipogenesis and accumulation of 4-hydroxynonenal in obese diabetic patients
Morana Jaganjac, Shamma Almuraikhy, Fatima Al-Khelaifi, Mashael Al-Jaber, Moataz Bashah, Nayef A. Mazloum, Kamelija Zarkovic, Neven Zarkovic, Georg Waeg, Wael Kafienah, Mohamed A. Elrayess
Redox Biology.2017; 12: 483. CrossRef - A Novel Index Using Soluble CD36 Is Associated with the Prevalence of Type 2 Diabetes Mellitus: Comparison Study with Triglyceride-Glucose Index
Ho Jin Kim, Jun Sung Moon, Il Rae Park, Joong Hee Kim, Ji Sung Yoon, Kyu Chang Won, Hyoung Woo Lee
Endocrinology and Metabolism.2017; 32(3): 375. CrossRef - Xanthine oxidase inhibition by febuxostat attenuates stress-induced hyperuricemia, glucose dysmetabolism, and prothrombotic state in mice
Maimaiti Yisireyili, Motoharu Hayashi, Hongxian Wu, Yasuhiro Uchida, Koji Yamamoto, Ryosuke Kikuchi, Mohammad Shoaib Hamrah, Takayuki Nakayama, Xian Wu Cheng, Tadashi Matsushita, Shigeo Nakamura, Toshimitsu Niwa, Toyoaki Murohara, Kyosuke Takeshita
Scientific Reports.2017;[Epub] CrossRef - Differential Effect of Sucrose and Fructose in Combination with a High Fat Diet on Intestinal Microbiota and Kidney Oxidative Stress
Adriana Rosas-Villegas, Mónica Sánchez-Tapia, Azalia Avila-Nava, Victoria Ramírez, Armando Tovar, Nimbe Torres
Nutrients.2017; 9(4): 393. CrossRef - Neovascular deterioration, impaired NADPH oxidase and inflammatory cytokine expression in adipose-derived multipotent cells from subjects with metabolic syndrome
Wilfredo Oliva-Olivera, Said Lhamyani, Leticia Coín-Aragüez, Daniel Castellano-Castillo, Juan Alcaide-Torres, Elena María Yubero-Serrano, Rajaa El Bekay, Francisco José Tinahones
Metabolism.2017; 71: 132. CrossRef - Fatty acid binding protein 4/aP2-dependent BLT1R expression and signaling
Ann V. Hertzel, Hongliang Xu, Michael Downey, Nicholas Kvalheim, David A. Bernlohr
Journal of Lipid Research.2017; 58(7): 1354. CrossRef - Metformin prevents glucotoxicity by alleviating oxidative and ER stress–induced CD36 expression in pancreatic beta cells
Jun Sung Moon, Udayakumar Karunakaran, Suma Elumalai, In-Kyu Lee, Hyoung Woo Lee, Yong-Woon Kim, Kyu Chang Won
Journal of Diabetes and its Complications.2017; 31(1): 21. CrossRef - Teneligliptin, a dipeptidyl peptidase-4 inhibitor, attenuated pro-inflammatory phenotype of perivascular adipose tissue and inhibited atherogenesis in normoglycemic apolipoprotein-E-deficient mice
Hotimah Masdan Salim, Daiju Fukuda, Yasutomi Higashikuni, Kimie Tanaka, Yoichiro Hirata, Shusuke Yagi, Takeshi Soeki, Michio Shimabukuro, Masataka Sata
Vascular Pharmacology.2017; 96-98: 19. CrossRef - Obesity-induced vascular dysfunction and arterial stiffening requires
endothelial cell arginase 1
Anil Bhatta, Lin Yao, Zhimin Xu, Haroldo A. Toque, Jijun Chen, Reem T. Atawia, Abdelrahman Y. Fouda, Zsolt Bagi, Rudolf Lucas, Ruth B. Caldwell, Robert W. Caldwell
Cardiovascular Research.2017; 113(13): 1664. CrossRef - Change in serum albumin concentration is inversely and independently associated with risk of incident metabolic syndrome
Sang-Man Jin, Yong Joo Hong, Jae Hwan Jee, Ji Cheol Bae, Kyu Yeon Hur, Moon-Kyu Lee, Jae Hyeon Kim
Metabolism.2016; 65(11): 1629. CrossRef
- The Relationship of Alcohol Consumption and Drinking Pattern to the Risk of Glomerular Hyperfiltration in Middle-aged Japanese Men: The Kansai Healthcare Study
- Obesity and Metabolic Syndrome
- Brown Fat and Browning for the Treatment of Obesity and Related Metabolic Disorders
- So Hun Kim, Jorge Plutzky
- Diabetes Metab J. 2016;40(1):12-21. Published online January 21, 2016
- DOI: https://doi.org/10.4093/dmj.2016.40.1.12
- 7,438 View
- 110 Download
- 155 Web of Science
- 158 Crossref
-
Abstract
PDFPubReader
Brown fat is a specialized fat depot that can increase energy expenditure and produce heat. After the recent discovery of the presence of active brown fat in human adults and novel transcription factors controlling brown adipocyte differentiation, the field of the study of brown fat has gained great interest and is rapidly growing. Brown fat expansion and/or activation results in increased energy expenditure and a negative energy balance in mice and limits weight gain. Brown fat is also able to utilize blood glucose and lipid and results in improved glucose metabolism and blood lipid independent of weight loss. Prolonged cold exposure and beta adrenergic agonists can induce browning of white adipose tissue. The inducible brown adipocyte, beige adipocyte evolving by thermogenic activation of white adipose tissue have different origin and molecular signature from classical brown adipocytes but share the characteristics of high mitochondria content, UCP1 expression and thermogenic capacity when activated. Increasing browning may also be an efficient way to increase whole brown fat activity. Recent human studies have shown possibilities that findings in mice can be reproduced in human, making brown fat a good candidate organ to treat obesity and its related disorders.
-
Citations
Citations to this article as recorded by- Modulation of angiogenic switch in reprogramming browning and lipid metabolism in white adipocytes
Sreelekshmi Sreekumar, Karyath Palliyath Gangaraj, Manikantan Syamala Kiran
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids.2024; 1869(1): 159423. CrossRef - Anti-Obesity Effects of GABA in C57BL/6J Mice with High-Fat Diet-Induced Obesity and 3T3-L1 Adipocytes
Heegu Jin, Hyein Han, Gunju Song, Hyun-Ji Oh, Boo-Yong Lee
International Journal of Molecular Sciences.2024; 25(2): 995. CrossRef - Citrus aurantium L. and synephrine improve brown adipose tissue function in adolescent mice programmed by early postnatal overfeeding
Andressa Cardoso Guimarães, Egberto Gaspar de Moura, Stephanie Giannini Silva, Bruna Pereira Lopes, Iala Milene Bertasso, Carla Bruna Pietrobon, Fernanda Torres Quitete, Tayanne de Oliveira Malafaia, Érica Patrícia Garcia Souza, Patrícia Cristina Lisboa,
Frontiers in Nutrition.2024;[Epub] CrossRef - Au@16-pH-16/miR-21 mimic nanosystem: An efficient treatment for obesity through browning and thermogenesis induction
Said Lhamyani, Adriana-Mariel Gentile, María Mengual-Mesa, Elia Grueso, Rosa M. Giráldez-Pérez, José Carlos Fernandez-Garcia, Antonio Vega-Rioja, Mercedes Clemente-Postigo, John R. Pearson, Isabel González-Mariscal, Gabriel Olveira, Francisco-Javier Bermu
Biomedicine & Pharmacotherapy.2024; 171: 116104. CrossRef - Walnut supplementation increases levels of UCP1 and CD36 in brown adipose tissue independently of diet type
Tamara Dakic, Dusan Jeremic, Iva Lakic, Nebojsa Jasnic, Aleksandra Ruzicic, Predrag Vujovic, Tanja Jevdjovic
Molecular and Cellular Biochemistry.2024;[Epub] CrossRef - Role of micro‐RNAs associated with adipose‐derived extracellular vesicles in metabolic disorders
Thomas Payet, Elisa Gabinaud, Jean‐François Landrier, Lourdes Mounien
Obesity Reviews.2024;[Epub] CrossRef - Spexin ameliorated obesity-related metabolic disorders through promoting white adipose browning mediated by JAK2-STAT3 pathway
Bihe Zeng, Qin Shen, Bo Wang, Xuan Tang, Jiaqi Jiang, Yiming Zheng, Hongbiao Huang, Wenyu Zhuo, Wang Wang, Yang Gao, Xuan Li, Shuhui Wang, Wenjie Li, Guanghui Qian, Jie Qin, Miao Hou, Haitao Lv
Nutrition & Metabolism.2024;[Epub] CrossRef - A Newly Synbiotic Combination Alleviates Obesity by Modulating the Gut Microbiota–Fat Axis and Inhibiting the Hepatic TLR4/NF‐κB Signaling Pathway
Yongbo Kang, Peng Ren, Xiaorong Shen, Xiaoyu Kuang, Xiaodan Yang, Haixia Liu, Huan Yan, Hao Yang, Xing Kang, Zeyuan Ding, Xuguang Luo, Jieqiong Ma, Ying Yang, Weiping Fan
Molecular Nutrition & Food Research.2023;[Epub] CrossRef - Lacticaseibacillus paracasei AO356 ameliorates obesity by regulating adipogenesis and thermogenesis in C57BL/6J male mice
Young In Kim, Eun-Sook Lee, Eun-Ji Song, Dong-Uk Shin, Ji-Eun Eom, Hee Soon Shin, Jung Eun Kim, Ju Yeoun Oh, Young-Do Nam, So-Young Lee
Journal of Functional Foods.2023; 101: 105404. CrossRef - Effects of caffeoylquinic acid analogs derived from aerial parts of Artemisia iwayomogi on adipogenesis
Su-Young Han, Jisu Kim, Bo Kyeong Kim, Wan Kyunn Whang, Hyeyoung Min
Food Science and Biotechnology.2023; 32(9): 1215. CrossRef - Anti-obesity effects of Lactiplantibacillus plantarum SKO-001 in high-fat diet-induced obese mice
Mi Jin Choi, Hana Yu, Jea Il Kim, Hee Seo, Ju Gyeong Kim, Seul-Ki Kim, Hak Sung Lee, Hyae Gyeong Cheon
European Journal of Nutrition.2023; 62(4): 1611. CrossRef - Meteorin-like Protein and Asprosin Levels in Children and Adolescents with Obesity and Their Relationship with Insulin Resistance and Metabolic Syndrome
Nariman Moradi, Reza Fadaei, Maryam Roozbehkia, Mitra Nourbakhsh, Mona Nourbakhsh, Maryam Razzaghy-Azar, Bagher Larijani
Laboratory Medicine.2023; 54(5): 457. CrossRef - A Novel Mix of Polyphenols and Micronutrients Reduces Adipogenesis and Promotes White Adipose Tissue Browning via UCP1 Expression and AMPK Activation
Francesca Pacifici, Gina Malatesta, Caterina Mammi, Donatella Pastore, Vincenzo Marzolla, Camillo Ricordi, Francesca Chiereghin, Marco Infante, Giulia Donadel, Francesco Curcio, Annalisa Noce, Valentina Rovella, Davide Lauro, Manfredi Tesauro, Nicola Di D
Cells.2023; 12(5): 714. CrossRef - Thromboxane A2-TP axis promotes adipose tissue macrophages M1 polarization leading to insulin resistance in obesity
Ruijie Xu, Yufeng Dai, Xu Zheng, Yongheng Yan, Zhao He, Hao Zhang, Haitao Li, Wei Chen
Biochemical Pharmacology.2023; 210: 115465. CrossRef - Green Tea Epigallocatechin Gallate Inhibits Preadipocyte Growth via the microRNA‐let‐7a/HMGA2 Signaling Pathway
Wen‐Ting Chen, Meei‐Ju Yang, Yi‐Wei Tsuei, Tsung‐Chen Su, An‐Ci Siao, Yow‐Chii Kuo, Ling‐Ru Huang, Yi Chen, Sy‐Jou Chen, Po‐Chuan Chen, Ching‐Feng Cheng, Hui‐Chen Ku, Yung‐Hsi Kao
Molecular Nutrition & Food Research.2023;[Epub] CrossRef - Current Role and Potential of Polymeric Biomaterials in Clinical Obesity Treatment
Rui-Chian Tang, I-Hsuan Yang, Feng-Huei Lin
Biomacromolecules.2023; 24(8): 3438. CrossRef - Short-duration cold exposure decreases fasting-induced glucose intolerance but has no effect on resting energy expenditure
Rima Solianik, Katerina Židonienė, Marius Brazaitis
Cryobiology.2023; 113: 104564. CrossRef - Caffeine promotes the production of Irisin in muscles and thus facilitates the browning of white adipose tissue
Chang Liu, Yi Li, Ge Song, Xuehan Li, Songyue Chen, Dixin Zou, Huixin Li, Chengyi Hu, Haotian Zhao, Yi Yan
Journal of Functional Foods.2023; 108: 105702. CrossRef - Thermogenic Modulation of Adipose Depots: A Perspective on Possible Therapeutic Intervention with Early Cardiorenal Complications of Metabolic Impairment
Ahmed F. El-Yazbi, Mohamed A. Elrewiny, Hosam M. Habib, Ali H. Eid, Perihan A. Elzahhar, Ahmed S.F. Belal
Molecular Pharmacology.2023; 104(5): 187. CrossRef - Rabbit Meat Extract Induces Browning in 3T3−L1 Adipocytes via the AMP−Activated Protein Kinase Pathway
In-Seon Bae, Jeong Ah Lee, Soo-Hyun Cho, Hyoun-Wook Kim, Yunseok Kim, Kangmin Seo, Hyun-Woo Cho, Min Young Lee, Ju Lan Chun, Ki Hyun Kim
Foods.2023; 12(19): 3671. CrossRef - Metabolic-Dysfunction-Associated Steatotic Liver Disease—Its Pathophysiology, Association with Atherosclerosis and Cardiovascular Disease, and Treatments
Hidekatsu Yanai, Hiroki Adachi, Mariko Hakoshima, Sakura Iida, Hisayuki Katsuyama
International Journal of Molecular Sciences.2023; 24(20): 15473. CrossRef - Brown Fat and Nutrition: Implications for Nutritional Interventions
Lloyd Noriega, Cheng-Ying Yang, Chih-Hao Wang
Nutrients.2023; 15(18): 4072. CrossRef - Metabolic Syndrome: A Narrative Review from the Oxidative Stress to the Management of Related Diseases
Giovanni Martemucci, Giuseppe Fracchiolla, Marilena Muraglia, Roberta Tardugno, Roberta Savina Dibenedetto, Angela Gabriella D’Alessandro
Antioxidants.2023; 12(12): 2091. CrossRef - Metabolic responses of light and taste receptors – unexpected actions of GPCRs in adipocytes
Onyinye Nuella Ekechukwu, Mark Christian
Reviews in Endocrine and Metabolic Disorders.2022; 23(1): 111. CrossRef - The ATF3 inducer protects against diet-induced obesity via suppressing adipocyte adipogenesis and promoting lipolysis and browning
Hui-Chen Ku, Tsai-Yun Chan, Jia-Fang Chung, Yung-Hsi Kao, Ching-Feng Cheng
Biomedicine & Pharmacotherapy.2022; 145: 112440. CrossRef - Prednisone stimulates white adipocyte browning via β3-AR/p38 MAPK/ERK signaling pathway
Sulagna Mukherjee, Jong Won Yun
Life Sciences.2022; 288: 120204. CrossRef - Tangeretin prevents obesity by modulating systemic inflammation, fat browning, and gut microbiota in high-fat diet-induced obese C57BL/6 mice
Qiyang Chen, Dan Wang, Yue Gu, Zixiao Jiang, Zhiqin Zhou
The Journal of Nutritional Biochemistry.2022; 101: 108943. CrossRef - Evaluation of thermal sensitivity is of potential clinical utility for the predictive, preventive, and personalized approach advancing metabolic syndrome management
Sujeong Mun, Kihyun Park, Siwoo Lee
EPMA Journal.2022; 13(1): 125. CrossRef - Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study
Hidekatsu Yanai, Hisayuki Katsuyama, Mariko Hakoshima
Biomedicines.2022; 10(2): 401. CrossRef - Historical and cultural aspects of obesity: From a symbol of wealth and prosperity to the epidemic of the 21st century
Marta Sumińska, Rafał Podgórski, Klaudia Bogusz‐Górna, Bogda Skowrońska, Artur Mazur, Marta Fichna
Obesity Reviews.2022;[Epub] CrossRef - Molecular Biological and Clinical Understanding of the Statin Residual Cardiovascular Disease Risk and Peroxisome Proliferator-Activated Receptor Alpha Agonists and Ezetimibe for Its Treatment
Hidekatsu Yanai, Hiroki Adachi, Mariko Hakoshima, Hisayuki Katsuyama
International Journal of Molecular Sciences.2022; 23(7): 3418. CrossRef - Pep19 Has a Positive Effect on Insulin Sensitivity and Ameliorates Both Hepatic and Adipose Tissue Phenotype of Diet-Induced Obese Mice
Renata Silvério, Robson Barth, Andrea S. Heimann, Patrícia Reckziegel, Gustavo J. dos Santos, Silvana Y. Romero-Zerbo, Francisco J. Bermúdez-Silva, Alex Rafacho, Emer S. Ferro
International Journal of Molecular Sciences.2022; 23(8): 4082. CrossRef - Thermogenic T cells: a cell therapy for obesity?
Ulf H. Beier, Daniel J. Baker, Joseph A. Baur
American Journal of Physiology-Cell Physiology.2022; 322(6): C1085. CrossRef - Accumulation of γδ T cells in visceral fat with aging promotes chronic inflammation
Maria E. C. Bruno, Sujata Mukherjee, Whitney L. Powell, Stephanie F. Mori, Franklyn K. Wallace, Beverly K. Balasuriya, Leon C. Su, Arnold J. Stromberg, Donald A. Cohen, Marlene E. Starr
GeroScience.2022; 44(3): 1761. CrossRef - Deletion of GPR30 Drives the Activation of Mitochondrial Uncoupling Respiration to Induce Adipose Thermogenesis in Female Mice
Jing Luo, Yao Wang, Elizabeth Gilbert, Dongmin Liu
Frontiers in Endocrinology.2022;[Epub] CrossRef - Inhibition of STAT3 enhances UCP1 expression and mitochondrial function in brown adipocytes
Lini Song, Xi Cao, Wenyi Ji, Lili Zhao, Weili Yang, Ming Lu, Jinkui Yang
European Journal of Pharmacology.2022; 926: 175040. CrossRef - TRPC5 deletion in the central amygdala antagonizes high-fat diet-induced obesity by increasing sympathetic innervation
Huan Ma, Chengkang He, Li Li, Peng Gao, Zongshi Lu, Yingru Hu, Lijuan Wang, Yu Zhao, Tingbing Cao, Yuanting Cui, Hongting Zheng, Gangyi Yang, Zhencheng Yan, Daoyan Liu, Zhiming Zhu
International Journal of Obesity.2022; 46(8): 1544. CrossRef - Adapalene induces adipose browning through the RARβ-p38 MAPK-ATF2 pathway
Na Hyun Lee, Mi Jin Choi, Hana Yu, Jea Il Kim, Hyae Gyeong Cheon
Archives of Pharmacal Research.2022; 45(5): 340. CrossRef - Engineering Functional Vascularized Beige Adipose Tissue from Microvascular Fragments of Models of Healthy and Type II Diabetes Conditions
Francisca M. Acosta, Katerina Stojkova, Jingruo Zhang, Eric Ivan Garcia Huitron, Jean X. Jiang, Christopher R. Rathbone, Eric M. Brey
Journal of Tissue Engineering.2022; 13: 204173142211093. CrossRef - Atractylenolide III from Atractylodes macrocephala Koidz promotes the activation of brown and white adipose tissue through SIRT1/PGC-1α signaling pathway
Xin Liu, Yuan Huang, Xu Liang, Qiong Wu, Nan Wang, Li-jun Zhou, Wen-wu Liu, Qun Ma, Bei Hu, Huan Gao, Ya-ling Cui, Xiang Li, Qing-chun Zhao
Phytomedicine.2022; 104: 154289. CrossRef - The pathobiology of perivascular adipose tissue (PVAT), the fourth layer of the blood vessel wall
Cassie Hillock-Watling, Avrum I. Gotlieb
Cardiovascular Pathology.2022; 61: 107459. CrossRef - RNA-Binding Proteins in the Regulation of Adipogenesis and Adipose Function
Pengpeng Zhang, Wenyan Wu, Chaofeng Ma, Chunyu Du, Yueru Huang, Haixia Xu, Cencen Li, Xiaofang Cheng, Ruijie Hao, Yongjie Xu
Cells.2022; 11(15): 2357. CrossRef - The Effect of Swimming Training with Cinnamon Consumption on β3-AR and ERK2 Gene Expression in the Visceral Adipose Tissue of Diabetic Rats
Kianoosh Mohammadi, Ali Khajehlandi, Amin Mohammadi
Gene, Cell and Tissue.2022;[Epub] CrossRef - Of mice and men: Considerations on adipose tissue physiology in animal models of obesity and human studies
Ioannis G. Lempesis, Dimitrios Tsilingiris, Junli Liu, Maria Dalamaga
Metabolism Open.2022; 15: 100208. CrossRef - The comparison of the effect of acute moderate and high-intensity exercise on the uncoupling protein -1 secretion
Desiana Merawati, Sugiharto Sugiharto, Adi Pranoto, Olivia Andiana, Prayogi Dwina Angga
Jurnal SPORTIF : Jurnal Penelitian Pembelajaran.2022; 8(2): 201. CrossRef - Omega-3 Polyunsaturated Fatty Acids Protect against High-Fat Diet-Induced Morphological and Functional Impairments of Brown Fat in Transgenic Fat-1 Mice
Lei Hao, Yong-Hui Nie, Chih-Yu Chen, Xiang-Yong Li, Kanakaraju Kaliannan, Jing X. Kang
International Journal of Molecular Sciences.2022; 23(19): 11903. CrossRef - Modulation of hypothalamic AMPK phosphorylation by olanzapine controls energy balance and body weight
Vitor Ferreira, Cintia Folgueira, Maria Guillén, Pablo Zubiaur, Marcos Navares, Assel Sarsenbayeva, Pilar López-Larrubia, Jan W. Eriksson, Maria J. Pereira, Francisco Abad-Santos, Guadalupe Sabio, Patricia Rada, Ángela M. Valverde
Metabolism.2022; 137: 155335. CrossRef - Okra (Abelmoschus esculentus L. Moench) prevents obesity by reducing lipid accumulation and increasing white adipose browning in high-fat diet-fed mice
Heegu Jin, Hyun-Ji Oh, Sehaeng Cho, Ok-Hwan Lee, Boo-Yong Lee
Food & Function.2022; 13(22): 11840. CrossRef - Functional Attenuation of UCP1 as the Potential Mechanism for a Thickened Blubber Layer in Cetaceans
Ming Zhou, Tianzhen Wu, Yue Chen, Shixia Xu, Guang Yang, Anne Yoder
Molecular Biology and Evolution.2022;[Epub] CrossRef - Brown adipose tissue transplantation as a novel alternative to obesity treatment: a systematic review
Moloud Payab, Mina Abedi, Najmeh Foroughi Heravani, Mahdieh Hadavandkhani, Maryam Arabi, Akram Tayanloo-Beik, Motahareh Sheikh Hosseini, Hadis Gerami, Fateme Khatami, Bagher Larijani, Mohammad Abdollahi, Babak Arjmand
International Journal of Obesity.2021; 45(1): 109. CrossRef - Corylin reduces obesity and insulin resistance and promotes adipose tissue browning through SIRT-1 and β3-AR activation
Chin-Chuan Chen, Chen-Hsin Kuo, Yann-Lii Leu, Shu-Huei Wang
Pharmacological Research.2021; 164: 105291. CrossRef - Inducible beige adipocytes improve impaired glucose metabolism in interscapular BAT-removal mice
Xiao-Wei Jia, Dong-Liang Fang, Xin-Yi Shi, Tao Lu, Chun Yang, Yan Gao
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids.2021; 1866(3): 158871. CrossRef - NNT‐induced tumor cell “slimming” reverses the pro‐carcinogenesis effect of HIF2a in tumors
Zhiyong Xiong, Wei Xiong, Wen Xiao, Changfei Yuan, Jian Shi, Yu Huang, Cheng Wang, Xiangui Meng, Zhixian Chen, Hongmei Yang, Ke Chen, Xiaoping Zhang
Clinical and Translational Medicine.2021;[Epub] CrossRef - CD10 marks non-canonical PPARγ-independent adipocyte maturation and browning potential of adipose-derived stem cells
Smarajit Chakraborty, Wee Kiat Ong, Winifred W. Y. Yau, Zhihong Zhou, K. N. Bhanu Prakash, Sue-Anne Toh, Weiping Han, Paul M. Yen, Shigeki Sugii
Stem Cell Research & Therapy.2021;[Epub] CrossRef - Regulatory roles of G-protein coupled receptors in adipose tissue metabolism and their therapeutic potential
Hyeonyeong Im, Ji-Hyun Park, Seowoo Im, Juhyeong Han, Kyungmin Kim, Yun-Hee Lee
Archives of Pharmacal Research.2021; 44(2): 133. CrossRef - 3-N-butylphthalide protects against high-fat-diet-induced obesity in C57BL/6 mice and increases metabolism in lipid-accumulating cells
Kang-Yun Lu, Shinn-Zong Lin, Kingsley Theras Primus Dass, Wei-Ju Lin, Shih-Ping Liu, Horng-Jyh Harn
Biomedicine & Pharmacotherapy.2021; 139: 111687. CrossRef - Ellagic acid induces beige remodeling of white adipose tissue by controlling mitochondrial dynamics and SIRT3
Woo Yong Park, Jinbong Park, Kwang Seok Ahn, Hyun Jeong Kwak, Jae‐Young Um
The FASEB Journal.2021;[Epub] CrossRef - Cajanolactone A, a Stilbenoid From Cajanus canjan (L.) Millsp, Prevents High-Fat Diet-Induced Obesity via Suppressing Energy Intake
Zhuohui Luo, Jiawen Huang, Zhiping Li, Zhiwen Liu, Linchun Fu, Yingjie Hu, Xiaoling Shen
Frontiers in Pharmacology.2021;[Epub] CrossRef - Zinc and the Innovative Zinc-α2-Glycoprotein Adipokine Play an Important Role in Lipid Metabolism: A Critical Review
Michalina Banaszak, Ilona Górna, Juliusz Przysławski
Nutrients.2021; 13(6): 2023. CrossRef - Natural products in the management of obesity: Fundamental mechanisms and pharmacotherapy
Yinghan Chan, Sin Wi Ng, Joycelin Zhu Xin Tan, Gaurav Gupta, Poonam Negi, Lakshmi Thangavelu, Sri Renukadevi Balusamy, Haribalan Perumalsamy, Wei Hsum Yap, Sachin Kumar Singh, Vanni Caruso, Kamal Dua, Dinesh Kumar Chellappan
South African Journal of Botany.2021; 143: 176. CrossRef - Group IIA secreted phospholipase A2 (PLA2G2A) augments adipose tissue thermogenesis
Michael S. Kuefner, Erin Stephenson, Mladen Savikj, Heather S. Smallwood, Qingming Dong, Christine Payré, Gérard Lambeau, Edwards A. Park
The FASEB Journal.2021;[Epub] CrossRef - N-butylidenephthalide ameliorates high-fat diet-induced obesity in mice and promotes browning through adrenergic response/AMPK activation in mouse beige adipocytes
Kang-Yun Lu, Kingsley Theras Primus Dass, Shinn-Zong Lin, Yu-Hua Tseng, Shih-Ping Liu, Horng-Jyh Harn
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids.2021; 1866(12): 159033. CrossRef - Green tea aqueous extract (GTAE) prevents high‐fat diet‐induced obesity by activating fat browning
Jie Li, Qiyang Chen, Xiuming Zhai, Dan Wang, Yujia Hou, Min Tang
Food Science & Nutrition.2021; 9(12): 6548. CrossRef - Ipragliflozin, an SGLT2 Inhibitor, Ameliorates High-Fat Diet-Induced Metabolic Changes by Upregulating Energy Expenditure through Activation of the AMPK/ SIRT1 Pathway
Ji-Yeon Lee, Minyoung Lee, Ji Young Lee, Jaehyun Bae, Eugene Shin, Yong-ho Lee, Byung-Wan Lee, Eun Seok Kang, Bong-Soo Cha
Diabetes & Metabolism Journal.2021; 45(6): 921. CrossRef - The increase of uncoupling protein-1 expression after moderate intensity continuous exercises in obese females
Sugiharto, Banih Sakti Adji, Desiana Merawati, Adi Pranoto
Jurnal SPORTIF : Jurnal Penelitian Pembelajaran.2021; 7(2): 194. CrossRef - Effects of Royal Jelly and Tocotrienol Rich Fraction in obesity treatment of calorie-restricted obese rats: a focus on white fat browning properties and thermogenic capacity
Naimeh Mesri Alamdari, Pardis Irandoost, Neda Roshanravan, Mohammadreza Vafa, Mohammad Asghari Jafarabadi, Shahriar Alipour, Leila Roshangar, Mohammadreza Alivand, Farnaz Farsi, Farzad Shidfar
Nutrition & Metabolism.2020;[Epub] CrossRef - Metabolic Benefits of MicroRNA-22 Inhibition
Marc Thibonnier, Christine Esau
Nucleic Acid Therapeutics.2020; 30(2): 104. CrossRef - Grape pomace extract supplementation activates FNDC5/irisin in muscle and promotes white adipose browning in rats fed a high-fat diet
Cecilia Rodriguez Lanzi, Diahann J. Perdicaro, Julián Gambarte Tudela, Victoria Muscia, Ariel R. Fontana, Patricia I. Oteiza, Marcela A. Vazquez Prieto
Food & Function.2020; 11(2): 1537. CrossRef - Citrus aurantium L. polymethoxyflavones promote thermogenesis of brown and white adipose tissue in high-fat diet induced C57BL/6J mice
Guangning Kou, Yan Hu, Zixiao Jiang, Zhenqing Li, Peiyuan Li, Hongjie Song, Qiyang Chen, Zhiqin Zhou, Quanjun Lyu
Journal of Functional Foods.2020; 67: 103860. CrossRef - Beneficial effects of pomegranate peel extract treatment on anthropometry and body composition of overweight patients with diabetes mellitus type-2: A randomised clinical trial
Milkica Grabež, Ranko Škrbić, Miloš Stojiljković, Vesna Rudić-Grujić, Katarina Šavikin, Nebojša Menković, Gordana Zdunić, Nađa Vasiljević
Scripta Medica.2020; 51(1): 21. CrossRef - Milk fat globule membrane and its component phosphatidylcholine induce adipose browning both in vivo and in vitro
Tiange Li, Min Du, Hanning Wang, Xueying Mao
The Journal of Nutritional Biochemistry.2020; 81: 108372. CrossRef - Dynamic contrast‐enhanced MRI of brown and beige adipose tissues
Jadegoud Yaligar, Sanjay Kumar Verma, Venkatesh Gopalan, Rengaraj Anantharaj, Giang Thi Thu Le, Kavita Kaur, Karthik Mallilankaraman, MK Leow, SS Velan
Magnetic Resonance in Medicine.2020; 84(1): 384. CrossRef - Taurine Stimulates Thermoregulatory Genes in Brown Fat Tissue and Muscle without an Influence on Inguinal White Fat Tissue in a High-Fat Diet-Induced Obese Mouse Model
Kyoung Soo Kim, Hari Madhuri Doss, Hee-Jin Kim, Hyung-In Yang
Foods.2020; 9(6): 688. CrossRef - White adipose tissue browning in critical illness: A review of the evidence, mechanisms and future perspectives
Elham Alipoor, Mohammad Javad Hosseinzadeh‐Attar, Mahsa Rezaei, Shima Jazayeri, Marianne Chapman
Obesity Reviews.2020;[Epub] CrossRef - Metabolic and energetic benefits of microRNA-22 inhibition
Marc Thibonnier, Christine Esau, Sujoy Ghosh, Edward Wargent, Claire Stocker
BMJ Open Diabetes Research & Care.2020; 8(1): e001478. CrossRef - Apple polyphenols induce browning of white adipose tissue
Yuki Tamura, Shigeto Tomiya, Junya Takegaki, Karina Kouzaki, Arata Tsutaki, Koichi Nakazato
The Journal of Nutritional Biochemistry.2020; 77: 108299. CrossRef - Adipose TBX1 regulates β-adrenergic sensitivity in subcutaneous adipose tissue and thermogenic capacity in vivo
Kathleen R. Markan, Lauren K. Boland, Abdul Qaadir King-McAlpin, Kristin E. Claflin, Michael P. Leaman, Morgan K. Kemerling, Madison M. Stonewall, Brad A. Amendt, James A. Ankrum, Matthew J. Potthoff
Molecular Metabolism.2020; 36: 100965. CrossRef - A systematic review of the association of neuregulin 4, a brown fat–enriched secreted factor, with obesity and related metabolic disturbances
Helda Tutunchi, Alireza Ostadrahimi, Mohammad‐Javad Hosseinzadeh‐Attar, Mahsa Miryan, Majid Mobasseri, Mehrangiz Ebrahimi‐Mameghani
Obesity Reviews.2020;[Epub] CrossRef - Deficiency of glutathione peroxidase-1 and catalase attenuated diet-induced obesity and associated metabolic disorders
Hyung-Ran Kim, Eun-Jeong Choi, Jeong-Hae Kie, Joo-Ho Lee, Ju-Young Seoh
Acta Diabetologica.2020; 57(2): 151. CrossRef - Clinical Application Potential of Small Molecules that Induce Brown Adipose Tissue Thermogenesis by Improving Fat Metabolism
Kang-Yun Lu, Kingsley Theras Primus Dass, Sheng-Feng Tsai, Hong-Meng Chuang, Shinn-Zong Lin, Shih-Ping Liu, Horng-Jyh Harn
Cell Transplantation.2020; 29: 096368972092739. CrossRef - MiR‐92a regulates brown adipocytes differentiation, mitochondrial oxidative respiration, and heat generation by targeting SMAD7
Zhipin Zhang, Huixin Jiang, Xiang Li, Xiaomin Chen, Yihua Huang
Journal of Cellular Biochemistry.2020; 121(8-9): 3825. CrossRef - Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator–activated receptor α
Darren M. Gordon, Kari L. Neifer, Abdul-Rizaq Ali Hamoud, Charles F. Hawk, Andrea L. Nestor-Kalinoski, Scott A. Miruzzi, Michael P. Morran, Samuel O. Adeosun, Jeffrey G. Sarver, Paul W. Erhardt, Robert E. McCullumsmith, David E. Stec, Terry D. Hinds
Journal of Biological Chemistry.2020; 295(29): 9804. CrossRef - The Presence of Active Brown Adipose Tissue Determines Cold-Induced Energy Expenditure and Oxylipin Profiles in Humans
Oana C Kulterer, Laura Niederstaetter, Carsten T Herz, Alexander R Haug, Andrea Bileck, Dietmar Pils, Alexandra Kautzky-Willer, Christopher Gerner, Florian W Kiefer
The Journal of Clinical Endocrinology & Metabolism.2020; 105(7): 2203. CrossRef - Downregulation of osteopontin inhibits browning of white adipose tissues through PI3K-AKT pathway in C57BL / 6 mice
Yi Lu, Yuhong Xu, Wanwan Yuan, Mengxi Wang, Yumeng Zhou, Kai Chen, Qiren Huang
European Journal of Pharmacology.2020; 866: 172822. CrossRef - Angiotensin AT1 receptor antagonism by losartan stimulates adipocyte browning via induction of apelin
Dong Young Kim, Mi Jin Choi, Tae Kyung Ko, Na Hyun Lee, Ok-Hee Kim, Hyae Gyeong Cheon
Journal of Biological Chemistry.2020; 295(44): 14878. CrossRef - Isoliquiritigenin Enhances the Beige Adipocyte Potential of Adipose-Derived Stem Cells by JNK Inhibition
Hanbyeol Moon, Jung-Won Choi, Byeong-Wook Song, Il-Kwon Kim, Soyeon Lim, Seahyoung Lee, Ki-Chul Hwang, Sang Woo Kim
Molecules.2020; 25(23): 5660. CrossRef - In vitro evaluation of Hydrilla verticillata for anti-adipogenesis activity on 3T3 L1 cell lines
SPandi Prabha, S Sadhana, C Karthik, DG Caroline
Pharmacognosy Magazine.2020; 16(5): 498. CrossRef - Leptin Promotes White Adipocyte Browning by Inhibiting the Hh Signaling Pathway
Jie Wang, Jing Ge, Haigang Cao, Xiaoyu Zhang, Yuan Guo, Xiao Li, Bo Xia, Gongshe Yang, Xin’e Shi
Cells.2019; 8(4): 372. CrossRef - Impact of Gut Microbiota on Host Glycemic Control
Céline Gérard, Hubert Vidal
Frontiers in Endocrinology.2019;[Epub] CrossRef - Effect of resveratrol on adipokines and myokines involved in fat browning: Perspectives in healthy weight against obesity
Oh Yoen Kim, Ji Yeon Chung, Juhyun Song
Pharmacological Research.2019; 148: 104411. CrossRef - A comprehensive diagnostic approach to detect underlying causes of obesity in adults
Eline S. van der Valk, Erica L.T. van den Akker, Mesut Savas, Lotte Kleinendorst, Jenny A. Visser, Mieke M. Van Haelst, Arya M. Sharma, Elisabeth F.C. van Rossum
Obesity Reviews.2019; 20(6): 795. CrossRef - Prostacyclin: A major prostaglandin in the regulation of adipose tissue development
Mohammad Sharifur Rahman
Journal of Cellular Physiology.2019; 234(4): 3254. CrossRef - The expression of brown fat‐associated proteins in colorectal cancer and the relationship of uncoupling protein 1 with prognosis
Abdo Alnabulsi, Beatriz Cash, Yehfang Hu, Linda Silina, Ayham Alnabulsi, Graeme I. Murray
International Journal of Cancer.2019; 145(4): 1138. CrossRef - Garlic augments the functional and nutritional behavior of Doenjang, a traditional Korean fermented soybean paste
Ashutosh Bahuguna, Shruti Shukla, Jong Suk Lee, Vivek K. Bajpai, So-Young Kim, Yun Suk Huh, Young-Kyu Han, Myunghee Kim
Scientific Reports.2019;[Epub] CrossRef - Clomiphene promotes browning of white adipocytes via casein kinase-2 inhibition
Hyun-Jin Kim, Dong Young Kim, Hyae Gyeong Cheon
European Journal of Pharmacology.2019; 861: 172596. CrossRef - Neuroendocrine Regulation of Energy Metabolism Involving Different Types of Adipose Tissues
Qi Zhu, Bradley J. Glazier, Benjamin C. Hinkel, Jingyi Cao, Lin Liu, Chun Liang, Haifei Shi
International Journal of Molecular Sciences.2019; 20(11): 2707. CrossRef - Tumor Cell “Slimming” Regulates Tumor Progression through PLCL1/UCP1‐Mediated Lipid Browning
Zhiyong Xiong, Wen Xiao, Lin Bao, Wei Xiong, Haibing Xiao, Yan Qu, Changfei Yuan, Hailong Ruan, Qi Cao, Keshan Wang, Zhengshuai Song, Cheng Wang, Wenjun Hu, Zeyuan Ru, Junwei Tong, Gong Cheng, Tianbo Xu, Xiangui Meng, Jian Shi, Zhixian Chen, Hongmei Yang,
Advanced Science.2019;[Epub] CrossRef - Soyasaponin Ab alleviates postmenopausal obesity through browning of white adipose tissue
Han-Jun Kim, Eun-Ji Choi, Hyo Sung Kim, Chan-Woong Choi, Sik-Won Choi, Sun-Lim Kim, Woo-Duck Seo, Sun Hee Do
Journal of Functional Foods.2019; 57: 453. CrossRef - Beneficial Effects of Pomegranate Peel Extract and Probiotics on Pre-adipocyte Differentiation
Valeria Sorrenti, Cinzia Lucia Randazzo, Cinzia Caggia, Gabriele Ballistreri, Flora Valeria Romeo, Simona Fabroni, Nicolina Timpanaro, Marco Raffaele, Luca Vanella
Frontiers in Microbiology.2019;[Epub] CrossRef - Visfatin; a potential novel mediator of brown adipose tissue
Parmida Pourvali-Talatappeh, Elham Alipoor
Obesity Medicine.2019; 15: 100122. CrossRef - Strategies and methods for the correction of obesity and associated cardiovascular risk
T. Yu. Kuznetsova, M. A. Druzhilov, G. A. Chumakova, N. D. Veselovskaya
Russian Journal of Cardiology.2019; (4): 61. CrossRef - Polymeric Carriers for Controlled Drug Delivery in Obesity Treatment
Di Huang, Meng Deng, Shihuan Kuang
Trends in Endocrinology & Metabolism.2019; 30(12): 974. CrossRef - Tbx15 is required for adipocyte browning induced by adrenergic signaling pathway
Wei Sun, Xuemei Zhao, Zhengqi Wang, Yi Chu, Liufeng Mao, Shaoqiang Lin, Xuefei Gao, Yuna Song, Xiaoyan Hui, Shiqi Jia, Shibing Tang, Yong Xu, Aimin Xu, Kerry Loomes, Cunchuan Wang, Donghai Wu, Tao Nie
Molecular Metabolism.2019; 28: 48. CrossRef - Effects of caloric restriction on the expression of lipocalin-2 and its receptor in the brown adipose tissue of high-fat diet-fed mice
Kyung-Ah Park, Zhen Jin, Hyeong Seok An, Jong Youl Lee, Eun Ae Jeong, Eun Bee Choi, Kyung Eun Kim, Hyun Joo Shin, Jung Eun Lee, Gu Seob Roh
The Korean Journal of Physiology & Pharmacology.2019; 23(5): 335. CrossRef - Peripherally administered melanocortins induce mice fat browning and prevent obesity
Adriana R. Rodrigues, Maria J. Salazar, Sílvia Rocha-Rodrigues, Inês O. Gonçalves, Célia Cruz, Delminda Neves, Henrique Almeida, José Magalhães, Alexandra M. Gouveia
International Journal of Obesity.2019; 43(5): 1058. CrossRef - Chinese medicine Jinlida granules improve high-fat-diet induced metabolic disorders via activation of brown adipose tissue in mice
Hui Zhang, Yuanyuan Hao, Cong Wei, Bing Yao, Shen Liu, Hongru Zhou, Dan Huang, Chuanhai Zhang, Yiling Wu
Biomedicine & Pharmacotherapy.2019; 114: 108781. CrossRef - Effects of lobeglitazone, a novel thiazolidinedione, on adipose tissue remodeling and brown and beige adipose tissue development in db/db mice
G Kim, Y-h Lee, M R Yun, J-Y Lee, E G Shin, B-W Lee, E S Kang, B-S Cha
International Journal of Obesity.2018; 42(3): 542. CrossRef - Air Pollution Has a Significant Negative Impact on Intentional Efforts to Lose Weight: A Global Scale Analysis
Morena Ustulin, So Young Park, Sang Ouk Chin, Suk Chon, Jeong-taek Woo, Sang Youl Rhee
Diabetes & Metabolism Journal.2018; 42(4): 320. CrossRef - Phytol stimulates the browning of white adipocytes through the activation of AMP-activated protein kinase (AMPK) α in mice fed high-fat diet
Fenglin Zhang, Wei Ai, Xiaoquan Hu, Yingying Meng, Cong Yuan, Han Su, Lina Wang, Xiaotong Zhu, Ping Gao, Gang Shu, Qingyan Jiang, Songbo Wang
Food & Function.2018; 9(4): 2043. CrossRef - Nobiletin induces brown adipocyte-like phenotype and ameliorates stress in 3T3-L1 adipocytes
Jameel Lone, Hilal Ahmad Parray, Jong Won Yun
Biochimie.2018; 146: 97. CrossRef - Regular aerobic exercise correlates with reduced anxiety and incresed levels of irisin in brain and white adipose tissue
Nazan Uysal, Oguz Yuksel, Servet Kizildag, Zeynep Yuce, Hikmet Gumus, Aslı Karakilic, Guven Guvendi, Basar Koc, Sevim Kandis, Mehmet Ates
Neuroscience Letters.2018; 676: 92. CrossRef - The Root of Atractylodes macrocephala Koidzumi Prevents Obesity and Glucose Intolerance and Increases Energy Metabolism in Mice
Mi Song, Soo-Kyoung Lim, Jing-Hua Wang, Hojun Kim
International Journal of Molecular Sciences.2018; 19(1): 278. CrossRef - Differential actions of PPAR-α and PPAR-β/δ on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice
Tamiris Lima Rachid, Flavia Maria Silva-Veiga, Francielle Graus-Nunes, Isabele Bringhenti, Carlos Alberto Mandarim-de-Lacerda, Vanessa Souza-Mello, Nobuyuki Takahashi
PLOS ONE.2018; 13(1): e0191365. CrossRef - Coordination Among Lipid Droplets, Peroxisomes, and Mitochondria Regulates Energy Expenditure Through the CIDE-ATGL-PPARα Pathway in Adipocytes
Linkang Zhou, Miao Yu, Muhammad Arshad, Wenmin Wang, Ye Lu, Jingyi Gong, Yangnan Gu, Peng Li, Li Xu
Diabetes.2018; 67(10): 1935. CrossRef - Lithocholic Acid Improves the Survival of Drosophila Melanogaster
Stefanie Staats, Gerald Rimbach, Axel Kuenstner, Simon Graspeuntner, Jan Rupp, Hauke Busch, Christian Sina, Ignacio R. Ipharraguerre, Anika E. Wagner
Molecular Nutrition & Food Research.2018;[Epub] CrossRef - Preconditioning lessens high fat induced metabolic syndrome along with markers of increased metabolic capacity in muscle and adipose tissue
Songpei Li, Xiu Zhou, Eunjung Jo, Ali Mahzari, Sherouk Fouda, Dongli Li, Kun Zhang, Ji-Ming Ye
Bioscience Reports.2018;[Epub] CrossRef - Alternative mRNA Splicing in the Pathogenesis of Obesity
Chi-Ming Wong, Lu Xu, Mabel Yau
International Journal of Molecular Sciences.2018; 19(2): 632. CrossRef - Krüppel-Like Factors
Nina M. Pollak, Matthew Hoffman, Ira J. Goldberg, Konstantinos Drosatos
JACC: Basic to Translational Science.2018; 3(1): 132. CrossRef - Dietary n-3 long-chain polyunsaturated fatty acids upregulate energy dissipating metabolic pathways conveying anti-obesogenic effects in mice
Stefanie Worsch, Mathias Heikenwalder, Hans Hauner, Bernhard L. Bader
Nutrition & Metabolism.2018;[Epub] CrossRef - Combination of Capsaicin and Hesperidin Reduces the Effectiveness of Each Compound To Decrease the Adipocyte Size and To Induce Browning Features in Adipose Tissue of Western Diet Fed Rats
Andrea Mosqueda-Solís, Juana Sánchez, María P. Portillo, Andreu Palou, Catalina Picó
Journal of Agricultural and Food Chemistry.2018; 66(37): 9679. CrossRef - Regulation of Adipose Tissue Metabolism by the Endocannabinoid System
Robin van Eenige, Mario van der Stelt, Patrick C.N. Rensen, Sander Kooijman
Trends in Endocrinology & Metabolism.2018; 29(5): 326. CrossRef - Cardiovascular and Metabolic Heterogeneity of Obesity
Ian J. Neeland, Paul Poirier, Jean-Pierre Després
Circulation.2018; 137(13): 1391. CrossRef - Zinc alpha2 glycoprotein promotes browning in adipocytes
Xin-Hua Xiao, Xiao-Yan Qi, Ya-Di Wang, Li Ran, Jing Yang, Huan-Li Zhang, Can-Xin Xu, Ge-Bo Wen, Jiang-Hua Liu
Biochemical and Biophysical Research Communications.2018; 496(2): 287. CrossRef - Impacts of High Sodium Intake on Obesity-related Gene Expression
Minjee Lee, Miyoung Park, Juhee Kim, Soyoung Sung, Myoungsook Lee
Journal of the East Asian Society of Dietary Life.2018; 28(5): 364. CrossRef - Programming mediated by fatty acids affects uncoupling protein 1 (UCP-1) in brown adipose tissue
Perla P. Argentato, Helena de Cássia César, Débora Estadella, Luciana P. Pisani
British Journal of Nutrition.2018; 120(6): 619. CrossRef - Reversal of Fatty Infiltration After Suprascapular Nerve Compression Release Is Dependent on UCP1 Expression in Mice
Zili Wang, Brian T. Feeley, Hubert T. Kim, Xuhui Liu
Clinical Orthopaedics & Related Research.2018; 476(8): 1665. CrossRef - Increase in relative skeletal muscle mass over time and its inverse association with metabolic syndrome development: a 7-year retrospective cohort study
Gyuri Kim, Seung-Eun Lee, Ji Eun Jun, You-Bin Lee, Jiyeon Ahn, Ji Cheol Bae, Sang-Man Jin, Kyu Yeon Hur, Jae Hwan Jee, Moon-Kyu Lee, Jae Hyeon Kim
Cardiovascular Diabetology.2018;[Epub] CrossRef - Myricetin-induced brown adipose tissue activation prevents obesity and insulin resistance in db/db mice
Tao Hu, Xiaoxue Yuan, Gang Wei, Haoshu Luo, Hyuek Jong Lee, Wanzhu Jin
European Journal of Nutrition.2018; 57(1): 391. CrossRef - Effects of growth hormone on uncoupling protein 1 in white adipose tissues in obese mice
Misa Hayashi, Kumi Futawaka, Rie Koyama, Yue Fan, Midori Matsushita, Asuka Hirao, Yuki Fukuda, Ayako Nushida, Syoko Nezu, Tetsuya Tagami, Kenji Moriyama
Growth Hormone & IGF Research.2017; 37: 31. CrossRef - Position paper of the European Society of Cardiology–working group of coronary pathophysiology and microcirculation: obesity and heart disease
Lina Badimon, Raffaele Bugiardini, Edina Cenko, Judit Cubedo, Maria Dorobantu, Dirk J. Duncker, Ramón Estruch, Davor Milicic, Dimitris Tousoulis, Zorana Vasiljevic, Gemma Vilahur, Cor de Wit, Akos Koller
European Heart Journal.2017; 38(25): 1951. CrossRef - Browning of Abdominal Aorta Perivascular Adipose Tissue Inhibits Adipose Tissue Inflammation
Run-Mei Li, Sui-Qing Chen, Ning-Xi Zeng, Shu-Hui Zheng, Li Guan, Hai-Mei Liu, Le-Quan Zhou, Jin-Wen Xu
Metabolic Syndrome and Related Disorders.2017; 15(9): 450. CrossRef - The role of adipose tissue in cancer-associated cachexia
Janina A Vaitkus, Francesco S Celi
Experimental Biology and Medicine.2017; 242(5): 473. CrossRef - Hormonal factors in the control of the browning of white adipose tissue
Jiamiao Hu, Mark Christian
Hormone Molecular Biology and Clinical Investigation.2017;[Epub] CrossRef - European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS)
Javier Egea, Isabel Fabregat, Yves M. Frapart, Pietro Ghezzi, Agnes Görlach, Thomas Kietzmann, Kateryna Kubaichuk, Ulla G. Knaus, Manuela G. Lopez, Gloria Olaso-Gonzalez, Andreas Petry, Rainer Schulz, Jose Vina, Paul Winyard, Kahina Abbas, Opeyemi S. Adem
Redox Biology.2017; 13: 94. CrossRef - Resveratrol enhances brown adipocyte formation and function by activating AMP‐activated protein kinase (AMPK) α1 in mice fed high‐fat diet
Songbo Wang, Xingwei Liang, Qiyuan Yang, Xing Fu, Meijun Zhu, B. D. Rodgers, Qingyan Jiang, Michael V. Dodson, Min Du
Molecular Nutrition & Food Research.2017;[Epub] CrossRef - Adipoz doku ve enerji metabolizması üzerine etkileri
Meltem Mermer, Nilüfer Acar TEK
SDÜ Sağlık Bilimleri Dergisi.2017;[Epub] CrossRef - Physiological roles of macrophages
Siamon Gordon, Luisa Martinez-Pomares
Pflügers Archiv - European Journal of Physiology.2017; 469(3-4): 365. CrossRef - Brown Adipose Tissue Bioenergetics: A New Methodological Approach
María Calderon‐Dominguez, Martín Alcalá, David Sebastián, Antonio Zorzano, Marta Viana, Dolors Serra, Laura Herrero
Advanced Science.2017;[Epub] CrossRef - Honokiol exerts dual effects on browning and apoptosis of adipocytes
Jameel Lone, Jong Won Yun
Pharmacological Reports.2017; 69(6): 1357. CrossRef - The Mixed Herbal Extract, CAPA, Prevents Obesity and Glucose Intolerance in Obese Mice
Miyoung Song
Journal of Korean Medicine for Obesity Research.2017; 17(2): 119. CrossRef - Brown Adipose Tissue: an Update on Recent Findings
Kara L. Marlatt, Eric Ravussin
Current Obesity Reports.2017; 6(4): 389. CrossRef - Radix Stellariae extract prevents high-fat-diet-induced obesity in C57BL/6 mice by accelerating energy metabolism
Yin Li, Xin Liu, Yu Fan, Baican Yang, Cheng Huang
PeerJ.2017; 5: e3305. CrossRef - Adipocyte arrestin domain-containing 3 protein (Arrdc3) regulates uncoupling protein 1 (Ucp1) expression in white adipose independently of canonical changes in β-adrenergic receptor signaling
Shannon H. Carroll, Ellen Zhang, Bing F. Wang, Katherine B. LeClair, Arifeen Rahman, David E. Cohen, Jorge Plutzky, Parth Patwari, Richard T. Lee, Makoto Kanzaki
PLOS ONE.2017; 12(3): e0173823. CrossRef - Effect of Long-Term Green Tea Extract Supplementation on Peripheral Blood Leukocytes in CrossFit-Trained and Untrained Men
Agata Żak, Ilona Pokora
Central European Journal of Sport Sciences and Medicine.2017; 19: 67. CrossRef - Rapid modulation of lipid metabolism in C57BL/6J mice induced by eicosapentaenoic acid-enriched phospholipid from Cucumaria frondosa
Lingyu Zhang, Dan Wang, Min Wen, Lei Du, Changhu Xue, Jingfeng Wang, Jie Xu, Yuming Wang
Journal of Functional Foods.2017; 28: 28. CrossRef - Egr1 deficiency induces browning of inguinal subcutaneous white adipose tissue in mice
Cécile Milet, Marianne Bléher, Kassandra Allbright, Mickael Orgeur, Fanny Coulpier, Delphine Duprez, Emmanuelle Havis
Scientific Reports.2017;[Epub] CrossRef - Purinergic Receptors in Adipose Tissue As Potential Targets in Metabolic Disorders
Marco Tozzi, Ivana Novak
Frontiers in Pharmacology.2017;[Epub] CrossRef - Phyllodulcin, a Natural Sweetener, Regulates Obesity-Related Metabolic Changes and Fat Browning-Related Genes of Subcutaneous White Adipose Tissue in High-Fat Diet-Induced Obese Mice
Eunju Kim, Soo-Min Lim, Min-Soo Kim, Sang-Ho Yoo, Yuri Kim
Nutrients.2017; 9(10): 1049. CrossRef - Yogurt and Cardiometabolic Diseases: A Critical Review of Potential Mechanisms
Melissa Anne Fernandez, Shirin Panahi, Noémie Daniel, Angelo Tremblay, André Marette
Advances in Nutrition.2017; 8(6): 812. CrossRef - Système endocannabinoïde : effets sur le métabolisme glucidique, mais aussi lipidique
A.-L. Sberna, P. Degrace, B. Vergès
Médecine des Maladies Métaboliques.2016; 10(5): 407. CrossRef - The Fat Side of the Endocannabinoid System: Role of Endocannabinoids in the Adipocyte
Isabelle Matias, Ilaria Belluomo, Daniela Cota
Cannabis and Cannabinoid Research.2016; 1(1): 176. CrossRef - Liraglutide suppresses obesity and induces brown fat-like phenotype via Soluble Guanylyl Cyclase mediated pathwayin vivoandin vitro
Endong Zhu, Yang Yang, Juanjuan Zhang, Yongmei Li, Chunjun Li, Liming Chen, Bei Sun
Oncotarget.2016; 7(49): 81077. CrossRef - NS5ATP6 modulates intracellular triglyceride content through FGF21 and independently of SIRT1 and SREBP1
Zhongshu Li, Shenghu Feng, Li Zhou, Shunai Liu, Jun Cheng
Biochemical and Biophysical Research Communications.2016; 475(1): 133. CrossRef - Novel device for male infertility screening with single-ball lens microscope and smartphone
Yoshitomo Kobori, Peter Pfanner, Gail S. Prins, Craig Niederberger
Fertility and Sterility.2016; 106(3): 574. CrossRef - An Intestinal Microbiota–Farnesoid X Receptor Axis Modulates Metabolic Disease
Frank J. Gonzalez, Changtao Jiang, Andrew D. Patterson
Gastroenterology.2016; 151(5): 845. CrossRef - Feast and famine: Adipose tissue adaptations for healthy aging
Daniele Lettieri Barbato, Katia Aquilano
Ageing Research Reviews.2016; 28: 85. CrossRef - Hypothalamus and thermogenesis: Heating the BAT, browning the WAT
Cristina Contreras, Rubén Nogueiras, Carlos Diéguez, Gema Medina-Gómez, Miguel López
Molecular and Cellular Endocrinology.2016; 438: 107. CrossRef - Concomitant beige adipocyte differentiation upon induction of mesenchymal stem cells into brown adipocytes
Yung-Li Wang, Shih-Pei Lin, Patrick C.H. Hsieh, Shih-Chieh Hung
Biochemical and Biophysical Research Communications.2016; 478(2): 689. CrossRef
- Modulation of angiogenic switch in reprogramming browning and lipid metabolism in white adipocytes
- Resistin in Rodents and Humans
- Hyeong Kyu Park, Rexford S. Ahima
- Diabetes Metab J. 2013;37(6):404-414. Published online December 12, 2013
- DOI: https://doi.org/10.4093/dmj.2013.37.6.404
- 5,584 View
- 52 Download
- 121 Crossref
-
Abstract
PDFPubReader
Obesity is characterized by excess accumulation of lipids in adipose tissue and other organs, and chronic inflammation associated with insulin resistance and an increased risk of type 2 diabetes. Obesity, type 2 diabetes, and cardiovascular diseases are major health concerns. Resistin was first discovered as an adipose-secreted hormone (adipokine) linked to obesity and insulin resistance in rodents. Adipocyte-derived resistin is increased in obese rodents and strongly related to insulin resistance. However, in contrast to rodents, resistin is expressed and secreted from macrophages in humans and is increased in inflammatory conditions. Some studies have also suggested an association between increased resistin levels and insulin resistance, diabetes and cardiovascular disease. Genetic studies have provided additional evidence for a role of resistin in insulin resistance and inflammation. Resistin appears to mediate the pathogenesis of atherosclerosis by promoting endothelial dysfunction, vascular smooth muscle cell proliferation, arterial inflammation, and formation of foam cells. Indeed, resistin is predictive of atherosclerosis and poor clinical outcomes in patients with coronary artery disease and ischemic stroke. There is also growing evidence that elevated resistin is associated with the development of heart failure. This review will focus on the biology of resistin in rodents and humans, and evidence linking resistin with type 2 diabetes, atherosclerosis, and cardiovascular disease.
-
Citations
Citations to this article as recorded by- Resistin – A Plausible Therapeutic Target in the Pathogenesis of Psoriasis
Manupati Srikanth, Mahaboobkhan Rasool
Immunological Investigations.2024; 53(2): 115. CrossRef - MHO or MUO? White adipose tissue remodeling
Jing Yi Zhao, Li Juan Zhou, Kai Le Ma, Rui Hao, Min Li
Obesity Reviews.2024;[Epub] CrossRef - Resistin in endocrine pancreas of sheep: Presence and expression related to different diets
Margherita Maranesi, Elisa Palmioli, Cecilia Dall'Aglio, Daniele Marini, Polina Anipchenko, Elena De Felice, Paola Scocco, Francesca Mercati
General and Comparative Endocrinology.2024; 348: 114452. CrossRef - Adipocytokines levels as potential biomarkers for discriminating patients with a diagnosis of depressive disorder from healthy controls
Elżbieta Małujło-Balcerska, Tadeusz Pietras
Journal of Psychiatric Research.2024; 171: 163. CrossRef - Adipokines in atopic dermatitis: the link between obesity and atopic dermatitis
Shiyun Zhang, Bingjie Zhang, Yuehua Liu, Li Li
Lipids in Health and Disease.2024;[Epub] CrossRef - The Role of Adipokines in the Control of Pituitary Functions
Barbara Kaminska, Beata Kurowicka, Marta Kiezun, Kamil Dobrzyn, Katarzyna Kisielewska, Marlena Gudelska, Grzegorz Kopij, Karolina Szymanska, Barbara Zarzecka, Oguzhan Koker, Ewa Zaobidna, Nina Smolinska, Tadeusz Kaminski
Animals.2024; 14(2): 353. CrossRef - Adipokine imbalance and its role in the pathogenesis of novel coronavirus infection
I. D. Bespalova, U. M. Mitrichenko, V. V. Kalyuzhin, E. S. Koroleva, Yu. I. Koshchavtseva, D. S. Romanov, D. E. Pershina
Bulletin of Siberian Medicine.2024; 22(4): 164. CrossRef - Association of adipokine levels with obesity in periodontal health and disease: A systematic review with meta‐analysis and meta‐regression
Eswar Kandaswamy, Chun‐Teh Lee, Soumya Bardvalli Gururaj, Sachin Shivanaikar, Vinayak M. Joshi
Journal of Periodontal Research.2024;[Epub] CrossRef - Association of maternal body composition and diet on breast milk hormones and neonatal growth during the first month of lactation
David Ramiro-Cortijo, Pratibha Singh, Gloria Herranz Carrillo, Andrea Gila-Díaz, María A. Martín-Cabrejas, Camilia R. Martin, Silvia M. Arribas
Frontiers in Endocrinology.2023;[Epub] CrossRef - Upregulation of peripheral blood mononuclear cells resistin gene expression in severe obstructive sleep apnea and obstructive sleep apnea with coexisting type 2 diabetes mellitus
Branislava Rajkov, Marija Zdravković, Ana Ninić, Milica Brajković, Slobodan Klašnja, Vera Gardijan, Lidija Memon, Jelena Munjas, Marija Mihajlović, Vesna Spasojević- Kalimanovska, Vojislav Radosavljević, Miron Sopić
Sleep and Breathing.2023; 27(5): 2031. CrossRef - Fat-to-heart crosstalk in health and disease
Fleur Lodewijks, Timothy A. McKinsey, Emma L. Robinson
Frontiers in Genetics.2023;[Epub] CrossRef - Adipokines as Diagnostic and Prognostic Markers for the Severity of COVID-19
Thomas Grewal, Christa Buechler
Biomedicines.2023; 11(5): 1302. CrossRef - Role of adipokines in sarcopenia
Wenhao Lu, Wenjie Feng, Jieyu Lai, Dongliang Yuan, Wenfeng Xiao, Yusheng Li
Chinese Medical Journal.2023; 136(15): 1794. CrossRef - Resistin, TNF-α, and microRNA 124-3p expressions in peripheral blood mononuclear cells are associated with diabetic nephropathy
Amin Monjezi, Azam Khedri, Mehrnoosh Zakerkish, Ghorban Mohammadzadeh
International Journal of Diabetes in Developing Countries.2022; 42(1): 62. CrossRef - Resistin in Urine and Breast Milk: Relation to Type of Feeding and Anthropometry at 1-Month
Irena Santosa, Hiromichi Shoji, Kentaro Awata, Yoshiteru Arai, Hiroki Suganuma, Toshiaki Shimizu
Pediatric Reports.2022; 14(1): 86. CrossRef - High Serum Levels of Resistin is Associated With Acute Cerebral Infarction
Kee Ook Lee, Kyung-Yul Lee, Cheol-Young Lee, Ji Hoon Kim, Jaeku Kang, Hoi Young Lee, Sang-Jun Na, Seung-Hun Oh, Ji Hoe Heo
The Neurologist.2022; 27(2): 41. CrossRef - Resistin production does not affect outcomes in a mouse model of acute surgical sepsis
Anthony S. Bonavia, Zissis C. Chroneos, Victor Ruiz-Velasco, Charles H. Lang, Partha Mukhopadhyay
PLOS ONE.2022; 17(3): e0265241. CrossRef - Single-nucleotide polymorphisms as important risk factors of diabetes among Middle East population
Iman Akhlaghipour, Amir Reza Bina, Mohammad Reza Mogharrabi, Ali Fanoodi, Amir Reza Ebrahimian, Soroush Khojasteh Kaffash, Atefeh Babazadeh Baghan, Mohammad Erfan Khorashadizadeh, Negin Taghehchian, Meysam Moghbeli
Human Genomics.2022;[Epub] CrossRef - Synergistic Effects of Weighted Genetic Risk Scores and Resistin and sST2 Levels on the Prognostication of Long-Term Outcomes in Patients with Coronary Artery Disease
Hsin-Hua Chou, Lung-An Hsu, Jyh-Ming Jimmy Juang, Fu-Tien Chiang, Ming-Sheng Teng, Semon Wu, Yu-Lin Ko
International Journal of Molecular Sciences.2022; 23(8): 4292. CrossRef - Hypoxia Increases the Potential for Neutrophil-mediated Endothelial Damage in Chronic Obstructive Pulmonary Disease
Katharine M. Lodge, Arlette Vassallo, Bin Liu, Merete Long, Zhen Tong, Paul R. Newby, Danya Agha-Jaffar, Koralia Paschalaki, Clara E. Green, Kylie B. R. Belchamber, Victoria C. Ridger, Robert A. Stockley, Elizabeth Sapey, Charlotte Summers, Andrew S. Cowb
American Journal of Respiratory and Critical Care Medicine.2022; 205(8): 903. CrossRef - The Role of the Adipokine Resistin in the Pathogenesis and Progression of Epithelial Ovarian Cancer
Klaudia Parafiniuk, Wiktoria Skiba, Anna Pawłowska, Dorota Suszczyk, Aleksandra Maciejczyk, Iwona Wertel
Biomedicines.2022; 10(4): 920. CrossRef - Resistin Modulates Low-Density Lipoprotein Cholesterol Uptake in Human Placental Explants via PCSK9
Sonia Nava-Salazar, Arturo Flores-Pliego, Giovanni Pérez-Martínez, Sandra Parra-Hernández, America Vanoye-Carlo, Francisco Ibarguengoitia-Ochoa, Otilia Perichart-Perera, Enrique Reyes-Muñoz, Juan Mario Solis-Paredes, Salvador Espino y Sosa, Guadalupe Estr
Reproductive Sciences.2022; 29(11): 3242. CrossRef - Differential Association of Selected Adipocytokines, Adiponectin, Leptin, Resistin, Visfatin and Chemerin, with the Pathogenesis and Progression of Type 2 Diabetes Mellitus (T2DM) in the Asir Region of Saudi Arabia: A Case Control Study
Mohammad Muzaffar Mir, Rashid Mir, Mushabab Ayed Abdullah Alghamdi, Javed Iqbal Wani, Zia Ul Sabah, Mohammed Jeelani, Vijaya Marakala, Shahzada Khalid Sohail, Mohamed O’haj, Muffarah Hamid Alharthi, Mohannad Mohammad S. Alamri
Journal of Personalized Medicine.2022; 12(5): 735. CrossRef - Immune system and sarcopenia: Presented relationship and future perspective
Xuzhi Zhang, Hengzhen Li, Miao He, Jingyu Wang, Yuxiang Wu, Yusheng Li
Experimental Gerontology.2022; 164: 111823. CrossRef - Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes
Giuseppina Biondi, Nicola Marrano, Anna Borrelli, Martina Rella, Giuseppe Palma, Isabella Calderoni, Edoardo Siciliano, Pasquale Lops, Francesco Giorgino, Annalisa Natalicchio
International Journal of Molecular Sciences.2022; 23(10): 5522. CrossRef - Supplemental hydroxychloroquine therapy regulates adipokines in patients with systemic lupus erythematosus with stable disease
Risa Wakiya, Kiyo Ueeda, Hiromi Shimada, Shusaku Nakashima, Tomohiro Kameda, Nobuyuki Miyatake, Mikiya Kato, Taichi Miyagi, Koichi Sugihara, Mao Mizusaki, Rina Mino, Norimitsu Kadowaki, Hiroaki Dobashi
Clinical Rheumatology.2022; 41(11): 3345. CrossRef - Can soy isoflavones in combination with soy protein change serum concentration of adiponectin and resistin? A systematic review and meta‐analysis on randomized clinical trials
Mitra Hariri, Bahareh Amirkalali, Ensiyeh Mollanoroozy, Ali Gholami
Food Science & Nutrition.2022; 10(12): 4126. CrossRef - Adipokines: Deciphering the cardiovascular signature of adipose tissue
Joseph C. Galley, Shubhnita Singh, Wanessa M.C. Awata, Juliano V. Alves, Thiago Bruder-Nascimento
Biochemical Pharmacology.2022; 206: 115324. CrossRef - Evaluation of the Anti-Obesity Effect of Zeaxanthin and Exercise in HFD-Induced Obese Rats
Mona Al-thepyani, Salha Algarni, Hana Gashlan, Mohamed Elzubier, Lina Baz
Nutrients.2022; 14(23): 4944. CrossRef - Single High-Dose Vitamin D Supplementation as an Approach for Reducing Ultramarathon-Induced Inflammation: A Double-Blind Randomized Controlled Trial
Jan Mieszkowski, Andżelika Borkowska, Błażej Stankiewicz, Andrzej Kochanowicz, Bartłomiej Niespodziński, Marcin Surmiak, Tomasz Waldziński, Rafał Rola, Miroslav Petr, Jędrzej Antosiewicz
Nutrients.2021; 13(4): 1280. CrossRef - Resistin mitigates stemness and metabolic profile of human adipose-derived mesenchymal stem cells via insulin resistance
Komal Rawal, Kishan M. Purohit, Tushar P. Patel, Neeta Karont, Sarita Gupta
Cytokine.2021; 138: 155374. CrossRef - Resistin is co-secreted with adiponectin in white mouse adipocytes
Saliha Musovic, Man Mohan Shrestha, Ali M. Komai, Charlotta S. Olofsson
Biochemical and Biophysical Research Communications.2021; 534: 707. CrossRef - Resistin: Potential biomarker and therapeutic target in atherosclerosis
Li Zhou, Jun-Yi Li, Ping-Ping He, Xiao-Hua Yu, Chao-Ke Tang
Clinica Chimica Acta.2021; 512: 84. CrossRef - The circulating levels of CTRP1 and CTRP5 are associated with obesity indices and carotid intima-media thickness (cIMT) value in patients with type 2 diabetes: a preliminary study
Ziba Majidi, Solaleh Emamgholipour, Abolfazl Omidifar, Soheil Rahmani Fard, Hossein Poustchi, Mehrnoosh Shanaki
Diabetology & Metabolic Syndrome.2021;[Epub] CrossRef - Corylin reduces obesity and insulin resistance and promotes adipose tissue browning through SIRT-1 and β3-AR activation
Chin-Chuan Chen, Chen-Hsin Kuo, Yann-Lii Leu, Shu-Huei Wang
Pharmacological Research.2021; 164: 105291. CrossRef - A Focused Review of the Metabolic Side-Effects of Clozapine
Jessica W. Y. Yuen, David D. Kim, Ric M. Procyshyn, William J. Panenka, William G. Honer, Alasdair M. Barr
Frontiers in Endocrinology.2021;[Epub] CrossRef - A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse
Antonio J. López-Gambero, Cristina Rosell-Valle, Dina Medina-Vera, Juan Antonio Navarro, Antonio Vargas, Patricia Rivera, Carlos Sanjuan, Fernando Rodríguez de Fonseca, Juan Suárez
International Journal of Molecular Sciences.2021; 22(10): 5365. CrossRef - Resistin in pregnancy: Analysis of determinants in pairs of umbilical cord blood and maternal serum
Anne Floeck, Nina Ferrari, Christine Joisten, Maria T. Puth, Brigitte Strizek, Ramona Dolscheid-Pommerich, Ulrich Gembruch, Waltraut M. Merz
Cytokine: X.2021; 3(2): 100052. CrossRef - Is resistin the master link between inflammation and inflammation-related chronic diseases?
Mohammed Taouis, Yacir Benomar
Molecular and Cellular Endocrinology.2021; 533: 111341. CrossRef - The dynamics of human bone marrow adipose tissue in response to feeding and fasting
Pouneh K. Fazeli, Miriam A. Bredella, Gisela Pachon-Peña, Wenxiu Zhao, Xun Zhang, Alexander T. Faje, Megi Resulaj, Sai P. Polineni, Tara M. Holmes, Hang Lee, Elizabeth K. O’Donnell, Ormond A. MacDougald, Mark C. Horowitz, Clifford J. Rosen, Anne Klibanski
JCI Insight.2021;[Epub] CrossRef - Resistin: A journey from metabolism to cancer
Ankita Deb, Bhavana Deshmukh, Pranay Ramteke, Firoz Khan Bhati, Manoj Kumar Bhat
Translational Oncology.2021; 14(10): 101178. CrossRef - Obesity is the basis of metabolic syndrome
A. F. Verbovoy, N. I. Verbovaya, Yu. A. Dolgikh
Obesity and metabolism.2021; 18(2): 142. CrossRef - Human Milk Metabolic Hormones: Analytical Methods and Current Understanding
Majed A. Suwaydi, Zoya Gridneva, Sharon L. Perrella, Mary E. Wlodek, Ching Tat Lai, Donna T. Geddes
International Journal of Molecular Sciences.2021; 22(16): 8708. CrossRef - Adipokines as Immune Cell Modulators in Multiple Sclerosis
Merel Rijnsburger, Niek Djuric, Inge A. Mulder, Helga E. de Vries
International Journal of Molecular Sciences.2021; 22(19): 10845. CrossRef - The Role of Adipokines in Cardiovascular Pathology
Valery Podzolkov , Anna Pokrovskaya, Ulyana Bazhanova , Tatyana Vargina , Svetlana Anatolievna Knyazeva , Daria Vanina
Open Access Macedonian Journal of Medical Sciences.2021; 9(F): 794. CrossRef - Measurement of Plasma Resistin Concentrations in Horses with Metabolic and Inflammatory Disorders
Beatriz Fuentes-Romero, Alberto Muñoz-Prieto, José J. Cerón, María Martín-Cuervo, Manuel Iglesias-García, Escolástico Aguilera-Tejero, Elisa Díez-Castro
Animals.2021; 12(1): 77. CrossRef - EFFECT OF DIET AND EXERCISE-INDUCE WEIGHT LOSS ON LEVEL OF RESISTIN IN PATIENT WITH OBESITY
О. I. Tokarenko, I. O. Andreieva, O. O. Tokarenko, M. M. Surmilo
Modern medical technology.2021; (4): 11. CrossRef - Alteration of gut microbiota affects expression of adiponectin and resistin through modifying DNA methylation in high-fat diet-induced obese mice
Hongyang Yao, Chaonan Fan, Yuanyuan Lu, Xiuqin Fan, Lulu Xia, Ping Li, Rui Wang, Tiantian Tang, Yuanyuan Wang, Kemin Qi
Genes & Nutrition.2020;[Epub] CrossRef - Resistin hormone in diabetic kidney disease and its relation to iron status and hepcidin
Zhian Sherzad Hayder, Zrar Saleem Kareem
International Urology and Nephrology.2020; 52(4): 749. CrossRef - Proteoglycans in Obesity-Associated Metabolic Dysfunction and Meta-Inflammation
Ariane R. Pessentheiner, G. Michelle Ducasa, Philip L. S. M. Gordts
Frontiers in Immunology.2020;[Epub] CrossRef - Resistin Is Increased in Periodontal Cells and Tissues: In Vitro and In Vivo Studies
Andressa V. B. Nogueira, Marjan Nokhbehsaim, Sema Tekin, Rafael S. de Molon, Luis C. Spolidorio, Svenja Memmert, Anna Damanaki, Andreas Jäger, Sigrun Eick, James Deschner, Joni A. Cirelli
Mediators of Inflammation.2020; 2020: 1. CrossRef - Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease
Alan Chait, Laura J. den Hartigh
Frontiers in Cardiovascular Medicine.2020;[Epub] CrossRef - The possible role of endocrine dysfunction of adipose tissue in gestational diabetes mellitus
Patrik Šimják, Kateřina Anderlová, Anna Cinkajzlová, Antonín Pařízek, Michal Kršek, Martin Haluzík
Minerva Endocrinologica.2020;[Epub] CrossRef - High Plasma Resistin Levels Portend the Insulin Resistance-Associated Susceptibility to Early Cognitive Decline in Patients with Type 2 Diabetes Mellitus
Chenchen Wang, Xi Huang, Sai Tian, Rong Huang, Dan Guo, Hongyan Lin, Jiaqi Wang, Shaohua Wang
Journal of Alzheimer's Disease.2020; 75(3): 807. CrossRef - Resistin in metabolism, inflammation, and disease
Deeksha Tripathi, Sashi Kant, Saurabh Pandey, Nasreen Z. Ehtesham
The FEBS Journal.2020; 287(15): 3141. CrossRef - Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases
Lucia Recinella, Giustino Orlando, Claudio Ferrante, Annalisa Chiavaroli, Luigi Brunetti, Sheila Leone
Frontiers in Physiology.2020;[Epub] CrossRef - Correlation of CCL18 with Levels of Adi-pokines in the Sera of Patients with Myocardial Infarction in a 6-Month Period: Case Series
Atefeh GamarTalepoor, Ehsan Dowlatshahi, Mehrnoush Doroudchi
Iranian South Medical Journal.2020; 23(3): 222. CrossRef - The Mesentery, Systemic Inflammation, and Crohn’s Disease
Edgardo D Rivera, John Calvin Coffey, Dara Walsh, Eli D Ehrenpreis
Inflammatory Bowel Diseases.2019; 25(2): 226. CrossRef - Resistin and adenylyl cyclase-associated protein 1 (CAP1) regulate the expression of genes related to insulin resistance in BNL CL.2 mouse liver cells
Dimiter Avtanski, Karin Chen, Leonid Poretsky
Data in Brief.2019; 25: 104112. CrossRef - Proteomic profile of patients with atrial fibrillation undergoing cardiac surgery†
Ilias P Doulamis, George Samanidis, Aspasia Tzani, Asier Antoranz, Anastasios Gkogkos, Panagiotis Konstantopoulos, Vaia Pliaka, Angeliki Minia, Leonidas G Alexopoulos, Despina N Perrea, Konstantinos Perreas
Interactive CardioVascular and Thoracic Surgery.2019; 28(1): 94. CrossRef - Angiotensin-(1-7), Adipokines and Inflammation
Deborah de Farias Lelis, Daniela Fernanda de Freitas, Amanda Souto Machado, Thaísa Soares Crespo, Sérgio Henrique Sousa Santos
Metabolism.2019; 95: 36. CrossRef - New Insights into Adipokines as Potential Biomarkers for Type-2 Diabetes Mellitus
Marta Olivera-Santa Catalina, Pedro C. Redondo, Maria P. Granados, Carlos Cantonero, Jose Sanchez-Collado, Letizia Albarran, Jose J. Lopez
Current Medicinal Chemistry.2019; 26(22): 4119. CrossRef - Myokine–adipokine cross-talk: potential mechanisms for the association between plasma irisin and adipokines and cardiometabolic risk factors in Mexican children with obesity and the metabolic syndrome
Adrian M. Gonzalez-Gil, Mariana Peschard-Franco, Elena C. Castillo, Gustavo Gutierrez-DelBosque, Victor Treviño, Christian Silva-Platas, Luisa Perez-Villarreal, Gerardo Garcia-Rivas, Leticia Elizondo-Montemayor
Diabetology & Metabolic Syndrome.2019;[Epub] CrossRef - Early Life Exposures to Perfluoroalkyl Substances in Relation to Adipokine Hormone Levels at Birth and During Childhood
Colleen Shelly, Philippe Grandjean, Youssef Oulhote, Peter Plomgaard, Ruth Frikke-Schmidt, Flemming Nielsen, Denis Zmirou-Navier, Pal Weihe, Damaskini Valvi
The Journal of Clinical Endocrinology & Metabolism.2019; 104(11): 5338. CrossRef - Overweight and obesity in childhood: Dietary, biochemical, inflammatory and lifestyle risk factors
Samah R. Albataineh, Eman F. Badran, Reema F. Tayyem
Obesity Medicine.2019; 15: 100112. CrossRef - Effects of major adipokines and the −420 C > G resistin gene polymorphism on the long-term outcome of patients with acute ischemic stroke
Stella Bouziana, Konstantinos Tziomalos, Antonis Goulas, Timoleon-Achilleas Vyzantiadis, Maria Papadopoulou, Athanasia Panderi, Apostolos Ι. Ηatzitolios
International Journal of Neuroscience.2019; 129(10): 978. CrossRef - The Complex Interactions Between Obesity, Metabolism and the Brain
Romina María Uranga, Jeffrey Neil Keller
Frontiers in Neuroscience.2019;[Epub] CrossRef - Resistin: A reappraisal
E. Acquarone, F. Monacelli, R. Borghi, A. Nencioni, P. Odetti
Mechanisms of Ageing and Development.2019; 178: 46. CrossRef - Implications of resistin in type 2 diabetes mellitus and coronary artery disease: Impairing insulin function and inducing pro‐inflammatory cytokines
Melissa Emamalipour, Khaled Seidi, Ali Jahanban‐Esfahlan, Rana Jahanban‐Esfahlan
Journal of Cellular Physiology.2019; 234(12): 21758. CrossRef - Serum-based soluble markers differentiate psoriatic arthritis from osteoarthritis
Vinod Chandran, Fatima Abji, Anthony V Perruccio, Rajiv Gandhi, Suzanne Li, Richard J Cook, Dafna D Gladman
Annals of the Rheumatic Diseases.2019; 78(6): 796. CrossRef - Telmisartan prevents diet-induced obesity and preserves leptin transport across the blood-brain barrier in high-fat diet-fed mice
Franziska Schuster, Gianna Huber, Ines Stölting, Emily E. Wing, Kathrin Saar, Norbert Hübner, William A. Banks, Walter Raasch
Pflügers Archiv - European Journal of Physiology.2018; 470(11): 1673. CrossRef - Adipokines in human breast milk
Juergen Kratzsch, Yoon Ju Bae, Wieland Kiess
Best Practice & Research Clinical Endocrinology & Metabolism.2018; 32(1): 27. CrossRef - Addressing the Perfect Storm: Biomarkers in Obesity and Pathophysiology of Cardiometabolic Risk
Krasimira Aleksandrova, Dariush Mozaffarian, Tobias Pischon
Clinical Chemistry.2018; 64(1): 142. CrossRef - Adipocytokine Involvement in Innate Immune Mechanisms
Paulina Żelechowska, Elżbieta Kozłowska, Joanna Pastwińska, Justyna Agier, Ewa Brzezińska-Błaszczyk
Journal of Interferon & Cytokine Research.2018; 38(12): 527. CrossRef - The effect of a garlic supplement on the pro-inflammatory adipocytokines, resistin and tumor necrosis factor-alpha, and on pain severity, in overweight or obese women with knee osteoarthritis
Sahar Dehghani, Elham Alipoor, Ahmad Salimzadeh, Mehdi Yaseri, Mostafa Hosseini, Christine Feinle-Bisset, Mohammad Javad Hosseinzadeh-Attar
Phytomedicine.2018; 48: 70. CrossRef - Perivascular adipose tissue (PVAT) in atherosclerosis: a double-edged sword
Xiao-Yan Qi, Shun-Lin Qu, Wen-Hao Xiong, Oren Rom, Lin Chang, Zhi-Sheng Jiang
Cardiovascular Diabetology.2018;[Epub] CrossRef - Usefulness of the Adipokines as Biomarkers of Ischemic Cardiac Dysfunction
Larisa-Diana Mocan Hognogi, Cerasela-Mihaela Goidescu, Anca-Daniela Farcaş
Disease Markers.2018; 2018: 1. CrossRef - Circulating fibroblast growth factor 21 in patients with liver cirrhosis
Sabrina Krautbauer, Lisa Rein-Fischboeck, Elisabeth M Haberl, Rebekka Pohl, Reiner Wiest, Christa Buechler
Clinical and Experimental Medicine.2018; 18(1): 63. CrossRef - Association of Cord Blood Resistin with Neonatal Birth Weight and Gestational Age
Shahnaz Pourarian, Saeed Fotouhikia, Forough Saki
Journal of Comprehensive Pediatrics.2018;[Epub] CrossRef - Major Adipokines and the −420C>G Resistin Gene Polymorphism as Predictors of Acute Ischemic Stroke Severity and In-Hospital Outcome
Styliani D. Bouziana, Konstantinos Tziomalos, Antonios Goulas, Timoleon-Achilleas Vyzantiadis, Athanasia Panderi, Apostolos Ι. Ηatzitolios
Journal of Stroke and Cerebrovascular Diseases.2018; 27(4): 963. CrossRef - Resistin and NGAL are associated with inflammatory response, endothelial activation and clinical outcomes in sepsis
Stephen P. J. Macdonald, Erika Bosio, Claire Neil, Glenn Arendts, Sally Burrows, Lisa Smart, Simon G. A. Brown, Daniel M. Fatovich
Inflammation Research.2017; 66(7): 611. CrossRef - Reference values for fasting serum resistin in healthy children and adolescents
Ulrik Lausten-Thomsen, Michael Christiansen, Paula Louise Hedley, Tenna Ruest Haarmark Nielsen, Cilius Esmann Fonvig, Oluf Pedersen, Torben Hansen, Jens-Christian Holm
Clinica Chimica Acta.2017; 469: 161. CrossRef - Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis
Alexander Kalinkovich, Gregory Livshits
Ageing Research Reviews.2017; 35: 200. CrossRef - Is There Any Relationship between Plasma 25-Hydroxyvitamin D3, Adipokine Profiles and Excessive Body Weight in Type 2 Diabetic Patients?
Joanna Kocot, Piotr Dziemidok, Małgorzata Kiełczykowska, Jacek Kurzepa, Grzegorz Szcześniak, Irena Musik
International Journal of Environmental Research and Public Health.2017; 15(1): 19. CrossRef - Exogenous Adipokine Peptide Resistin Protects Against Focal Cerebral Ischemia/Reperfusion Injury in Mice
Jiangtao Zhu, Di Wu, Chenyu Zhao, Man Luo, Ronald C. Hamdy, Balvin H. L. Chua, Xingshun Xu, Zhigang Miao
Neurochemical Research.2017; 42(10): 2949. CrossRef - Adipokines in Liver Cirrhosis
Christa Buechler, Elisabeth Haberl, Lisa Rein-Fischboeck, Charalampos Aslanidis
International Journal of Molecular Sciences.2017; 18(7): 1392. CrossRef - The role of sex steroids in white adipose tissue adipocyte function
A E Newell-Fugate
Reproduction.2017; 153(4): R133. CrossRef - Odanacatib Inhibits Resistin-induced Cardiomyocyte Hypertrophy Through the Inactivation of ERK Signaling Pathway
Xian Zheng, Guanchang Cheng, Jianwei Luo, Qunhui Ye, Yongzhi Deng, Lin Wu
International Journal of Pharmacology.2017; 13(2): 212. CrossRef - Linking resistin, inflammation, and cardiometabolic diseases
Hyeong Kyu Park, Mi Kyung Kwak, Hye Jeong Kim, Rexford S. Ahima
The Korean Journal of Internal Medicine.2017; 32(2): 239. CrossRef - Translating the biology of adipokines in atherosclerosis and cardiovascular diseases: Gaps and open questions
M. Ruscica, A. Baragetti, A.L. Catapano, G.D. Norata
Nutrition, Metabolism and Cardiovascular Diseases.2017; 27(5): 379. CrossRef - Differences in Mean Levels of Maternal Resistin Serum between Early Onset Preeclampsia (EOPE) and Late Onset Preeclampsia (LOPE)
Yusrawati ., P. Alfajra, R. Machmud
Research Journal of Obstetrics and Gynecology.2016; 10(1): 1. CrossRef - Secret talk between adipose tissue and central nervous system via secreted factors—an emerging frontier in the neurodegenerative research
Avinash Parimisetty, Anne-Claire Dorsemans, Rana Awada, Palaniyandi Ravanan, Nicolas Diotel, Christian Lefebvre d’Hellencourt
Journal of Neuroinflammation.2016;[Epub] CrossRef - The role of adipokines in ischemic stroke risk stratification
Styliani Bouziana, Konstantinos Tziomalos, Antonios Goulas, Apostolos Ι Ηatzitolios
International Journal of Stroke.2016; 11(4): 389. CrossRef - The endocrine function of human placenta: an overview
Mariana A. Costa
Reproductive BioMedicine Online.2016; 32(1): 14. CrossRef - Ursolic acid plays a protective role in obesity-induced cardiovascular diseases
Yu-Ting Lin, Ya-Mei Yu, Weng-Cheng Chang, Su-Yin Chiang, Hsu-Chin Chan, Ming-Fen Lee
Canadian Journal of Physiology and Pharmacology.2016; 94(6): 627. CrossRef - Determinants of body weight regulation in humans
Milene Moehlecke, Luis Henrique Canani, Lucas Oliveira Junqueira e Silva, Manoel Roberto Maciel Trindade, Rogerio Friedman, Cristiane Bauermann Leitão
Archives of Endocrinology and Metabolism.2016; 60(2): 152. CrossRef - Sitagliptin decreases ventricular arrhythmias by attenuated glucose-dependent insulinotropic polypeptide (GIP)-dependent resistin signalling in infarcted rats
Tsung-Ming Lee, Wei-Ting Chen, Nen-Chung Chang
Bioscience Reports.2016;[Epub] CrossRef - Les adipokines : état des lieux et nouveautés
J.-P. Bastard, C. Bastard, S. Fellahi, C. Vatier, J. Capeau, B. Fève
Obésité.2016; 11(3): 181. CrossRef - Factors that promote macrophage homing to adipose tissue in metabolic syndrome
Ishwarlal Jialal, Beverley Adams-Huet, Sridevi Devaraj
Journal of Diabetes and its Complications.2016; 30(8): 1434. CrossRef - Uncovering Factors Related to Pancreatic Beta-Cell Function
Aoife M. Curran, Miriam F. Ryan, Elaine Drummond, Eileen R. Gibney, Michael J. Gibney, Helen M. Roche, Lorraine Brennan, Nigel Irwin
PLOS ONE.2016; 11(8): e0161350. CrossRef - Resistin’s, obesity and insulin resistance: the continuing disconnect between rodents and humans
X. Huang, Z. Yang
Journal of Endocrinological Investigation.2016; 39(6): 607. CrossRef - Adipocytokines in renal transplant recipients
Kristof Nagy, Shankar Prasad Nagaraju, Connie M. Rhee, Zoltan Mathe, Miklos Z. Molnar
Clinical Kidney Journal.2016; 9(3): 359. CrossRef - Endocrine alterations from concentric vs. eccentric muscle actions: A brief review
Robert R. Kraemer, V. Daniel Castracane
Metabolism.2015; 64(2): 190. CrossRef - Non-traditional cytokines: How catecholamines and adipokines influence macrophages in immunity, metabolism and the central nervous system
Mark A. Barnes, Monica J. Carson, Meera G. Nair
Cytokine.2015; 72(2): 210. CrossRef - Local and serum levels of adipokines in patients with obesity after periodontal therapy: one‐year follow‐up
Tiago Eduardo Dias Gonçalves, Glaucia Santos Zimmermann, Luciene Cristina Figueiredo, Monique de Carvalho Souza, Daniele Ferreira da Cruz, Marta Ferreira Bastos, Hélio Doyle Pereira da Silva, Poliana Mendes Duarte
Journal of Clinical Periodontology.2015; 42(5): 431. CrossRef - Newborn Adipokines and Birth Outcomes
Edwina H. Yeung, Alexander C. McLain, Nancy Anderson, David Lawrence, Nansi S. Boghossian, Charlotte Druschel, Erin Bell
Paediatric and Perinatal Epidemiology.2015; 29(4): 317. CrossRef - The effect of a preparation of minerals, vitamins and trace elements on the cardiac gene expression pattern in male diabetic rats
Márta Sárközy, Gergő Szűcs, Márton Pipicz, Ágnes Zvara, Katalin Éder, Veronika Fekete, Csilla Szűcs, Judit Bárkányi, Csaba Csonka, László G. Puskás, Csaba Kónya, Péter Ferdinandy, Tamás Csont
Cardiovascular Diabetology.2015;[Epub] CrossRef - Diet-induced variability of the resistin gene (Retn) transcript level and methylation profile in rats
Joanna Nowacka-Woszuk, Ewa Pruszynska-Oszmalek, Maciej Szydlowski, Slawomir Sadkowski, Izabela Szczerbal
BMC Genetics.2015;[Epub] CrossRef - INFLUENCE OF RESISTIN ON THE COURSE OF ISCHEMIC HEART DISEASE IN PATIENTS WITH TYPE 2 DIABETES MELLITUS
A. T. Teplyakov, Sh. D. Akhmedov, T. Ye. Suslova, А. V. Andriyanova, A. V. Kuznetsova, N. V. Protopopova, V. V. Kalyuzhin, O. N. Nasanova
Bulletin of Siberian Medicine.2015; 14(5): 73. CrossRef - The Effects of a Single Developmentally Entrained Pulse of Testosterone in Female Neonatal Mice on Reproductive and Metabolic Functions in Adult Life
Hyeran Jang, Shalender Bhasin, Tyler Guarneri, Carlo Serra, Mary Schneider, Mi-Jeong Lee, Wen Guo, Susan K. Fried, Karol Pencina, Ravi Jasuja
Endocrinology.2015; 156(10): 3737. CrossRef - The Resin fromProtium heptaphyllumPrevents High-Fat Diet-Induced Obesity in Mice: Scientific Evidence and Potential Mechanisms
Karine Maria Martins Bezerra Carvalho, José Delano Barreto Marinho Filho, Tiago Sousa de Melo, Ana Jérsia Araújo, Josiane da Silva Quetz, Maria do Perpétuo Socorro Saldanha da Cunha, Karina Moura de Melo, Armenio Andre de Carvalho Almeida da Silva, Adrian
Evidence-Based Complementary and Alternative Medicine.2015; 2015: 1. CrossRef - Evolution of the Vertebrate Resistin Gene Family
Qingda Hu, Huanran Tan, David M. Irwin, Marc Robinson-Rechavi
PLOS ONE.2015; 10(6): e0130188. CrossRef - Obesity, adipokines and neuroinflammation
Argel Aguilar-Valles, Wataru Inoue, Christoph Rummel, Giamal N. Luheshi
Neuropharmacology.2015; 96: 124. CrossRef - Adipokines at the crossroad between obesity and cardiovascular disease
Filippo Molica, Sandrine Morel, Brenda Kwak, Françoise Rohner-Jeanrenaud, Sabine Steffens
Thrombosis and Haemostasis.2015; 113(03): 553. CrossRef - Resistin – 420 C/G polymorphism and serum resistin level in Iranian patients with gestational diabetes mellitus
Mohammad Ali Takhshid, Zinab Zare
Journal of Diabetes & Metabolic Disorders.2015;[Epub] CrossRef - Ictal adipokines are associated with pain severity and treatment response in episodic migraine
Nu Cindy Chai, Bizu Gelaye, Gretchen E. Tietjen, Paul D. Dash, Barbara A. Gower, Linda W. White, Thomas N. Ward, Ann I. Scher, B. Lee Peterlin
Neurology.2015; 84(14): 1409. CrossRef - Inflammation and insulin/IGF-1 resistance as the possible link between obesity and neurodegeneration
Lindsay J. Spielman, Jonathan P. Little, Andis Klegeris
Journal of Neuroimmunology.2014; 273(1-2): 8. CrossRef - Wild Blueberries (Vaccinium myrtillus) Alleviate Inflammation and Hypertension Associated with Developing Obesity in Mice Fed with a High-Fat Diet
Otto T. Mykkänen, Anne Huotari, Karl-Heinz Herzig, Thomas W. Dunlop, Hannu Mykkänen, Pirkka V. Kirjavainen, Michael Müller
PLoS ONE.2014; 9(12): e114790. CrossRef - Role of fat and adipokines in intestinal inflammation
LeaI Kredel, Arvind Batra, Britta Siegmund
Current Opinion in Gastroenterology.2014; 30(6): 559. CrossRef -
13C metabolic flux analysis shows that resistin impairs the metabolic response to insulin in L6E9 myotubes
Shirley Guzmán, Silvia Marin, Anibal Miranda, Vitaly A Selivanov, Josep J Centelles, Romain Harmancey, Fatima Smih, Annie Turkieh, Yves Durocher, Antonio Zorzano, Philippe Rouet, Marta Cascante
BMC Systems Biology.2014;[Epub] CrossRef - Bee Pollen Improves Muscle Protein and Energy Metabolism in Malnourished Old Rats through Interfering with the Mtor Signaling Pathway and Mitochondrial Activity
Jérôme Salles, Nicolas Cardinault, Véronique Patrac, Alexandre Berry, Christophe Giraudet, Marie-Laure Collin, Audrey Chanet, Camille Tagliaferri, Philippe Denis, Corinne Pouyet, Yves Boirie, Stéphane Walrand
Nutrients.2014; 6(12): 5500. CrossRef
- Resistin – A Plausible Therapeutic Target in the Pathogenesis of Psoriasis
- An
In Vitro Model to Probe the Regulation of Adipocyte Differentiation under Hyperglycemia - Kusampudi Shilpa, Thangaraj Dinesh, Baddireddi Subhadra Lakshmi
- Diabetes Metab J. 2013;37(3):176-180. Published online June 14, 2013
- DOI: https://doi.org/10.4093/dmj.2013.37.3.176
- 3,049 View
- 34 Download
- 8 Crossref
-
Abstract
PDFPubReader
Background The aim of this study was an
in vitro investigation of the effect of high glucose concentration on adipogenesis, as prolonged hyperglycemia alters adipocyte differentiation.Methods 3T3-L1 preadipocytes differentiated in the presence of varying concentrations of glucose (25, 45, 65, 85, and 105 mM) were assessed for adipogenesis using AdipoRed (Lonza) assay. Cell viability and proliferation were measured using MTT reduction and [3H] thymidine incorporation assay. The extent of glucose uptake and glycogen synthesis were measured using radiolabelled 2-deoxy-D-[1-3H] glucose and [14C]-UDP-glucose. The gene level expression was evaluated using reverse transcription-polymerase chain reaction and protein expression was studied using Western blot analysis.
Results Glucose at 105 mM concentration was observed to inhibit adipogenesis through inhibition of CCAAT-enhancer-binding proteins, sterol regulatory element-binding protein, peroxisome proliferator-activated receptor and adiponectin. High concentration of glucose induced stress by increasing levels of toll-like receptor 4, nuclear factor κB and tumor necrosis factor α thereby generating activated preadipocytes. These cells entered the state of hyperplasia through inhibition of p27 and proliferation was found to increase through activation of protein kinase B via phosphoinositide 3 kinase dependent pathway. This condition inhibited insulin signaling through decrease in insulin receptor β. Although the glucose transporter 4 (GLUT4) protein remained unaltered with the glycogen synthesis inhibited, the cells were found to exhibit an increase in glucose uptake via GLUT1.
Conclusion Adipogenesis in the presence of 105 mM glucose leads to an uncontrolled proliferation of activated preadipocytes providing an insight towards understanding obesity.
-
Citations
Citations to this article as recorded by- Adipogenesis-Related Metabolic Condition Affects Shear-Stressed Endothelial Cells Activity Responding to Titanium
Thaís Silva Pinto, Anderson Moreira Gomes, Paula Bertin de Morais, Willian F. Zambuzzi
Journal of Functional Biomaterials.2023; 14(3): 162. CrossRef - Chronic and Transient Hyperglycemia Induces Changes in the Expression Patterns of IL6 and ADIPOQ Genes and Their Associated Epigenetic Modifications in Differentiating Human Visceral Adipocytes
Adam Wróblewski, Justyna Strycharz, Ewa Świderska, Aneta Balcerczyk, Janusz Szemraj, Józef Drzewoski, Agnieszka Śliwińska
International Journal of Molecular Sciences.2021; 22(13): 6964. CrossRef - Effects of high glucose conditions on the expansion and differentiation capabilities of mesenchymal stromal cells derived from rat endosteal niche
Ahmed Makki A. Al-Qarakhli, Norhayati Yusop, Rachel J. Waddington, Ryan Moseley
BMC Molecular and Cell Biology.2019;[Epub] CrossRef - Inhibition of WNT/β-catenin signaling under serum starvation and hypoxia induces adipocytic transdifferentiation in human leiomyoma cells
Hiroshi Harada, Yojiro Tsuda, Kei Yabuki, Eisuke Shiba, Kazuyoshi Uchihashi, Atsuji Matsuyama, Yoshihisa Fujino, Toru Hachisuga, Masanori Hisaoka
Laboratory Investigation.2018; 98(4): 439. CrossRef - Effects of high glucose on caveolin-1 and insulin signaling in 3T3-L1 adipocytes
Sara Palacios-Ortega, Maider Varela-Guruceaga, J. Alfredo Martínez, Carlos de Miguel, Fermín I. Milagro
Adipocyte.2016; 5(1): 65. CrossRef - Pathophysiological role of enhanced bone marrow adipogenesis in diabetic complications
Meghan A Piccinin, Zia A Khan
Adipocyte.2014; 3(4): 263. CrossRef - Letter: AnIn VitroModel to Probe the Regulation of Adipocyte Differentiation under Hyperglycemia (Diabetes Metab J2013;37:176-80)
In-Kyung Jeong
Diabetes & Metabolism Journal.2013; 37(4): 296. CrossRef - Response: AnIn VitroModel to Probe the Regulation of Adipocyte Differentiation under Hyperglycemia (Diabetes Metab J2013;37:176-80)
Kusampudi Shilpa, Thangaraj Dinesh, Baddireddi Subhadra Lakshmi
Diabetes & Metabolism Journal.2013; 37(4): 298. CrossRef
- Adipogenesis-Related Metabolic Condition Affects Shear-Stressed Endothelial Cells Activity Responding to Titanium
 First
First Prev
Prev