- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 35(2); 2011 > Article
-
ReviewThe Importance of Global Studies of the Genetics of Type 2 Diabetes
- Mark I. McCarthy
-
Diabetes & Metabolism Journal 2011;35(2):91-100.
DOI: https://doi.org/10.4093/dmj.2011.35.2.91
Published online: April 30, 2011
Oxford Centre for Diabetes, Endocrinology and Metabolism, and Wellcome Trust Centre for Human Genetics, University of Oxford; Oxford NIHR Biomedical Research Centre, Churchill Hospital, Oxford, UK.
- Corresponding author: Mark I. McCarthy. Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital, Old Road, Headington, Oxford, OX3 7LJ, UK. mark.mccarthy@drl.ox.ac.uk
Copyright © 2011 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- EXPLORING THE OVERLAP IN ASSOCIATION SIGNALS BETWEEN MAJOR ETHNIC GROUPS
- TRANSETHNIC FINE MAPPING FOR COMMON VARIANT SIGNALS
- INFORMATION ON GENETIC ARCHITECTURE AND SELECTION
- EXPLAINING DIFFERENCES IN PREVALENCE AND PRESENTATION OF DISEASE
- ADVANCING BIOLOGICAL UNDERSTANDING OF DISEASE PREDISPOSITION
- THE FUTURE
- ACKNOWLEDGMENT
- REFERENCES
ABSTRACT
- Genome wide association analyses have revealed large numbers of common variants influencing predisposition to type 2 diabetes and related phenotypes. These studies have predominantly featured European populations, but are now being extended to samples from a wider range of ethnic groups. The transethnic analysis of association data is already providing insights into the genetic, molecular and biological causes of diabetes, and the relevance of such studies will increase as human discovery genetics increasingly moves towards sequencing-based approaches and a focus on low frequency and rare variants.
- The past few years have seen an explosion in our capacity to identify DNA sequence variants that influence individual predisposition to type 2 diabetes and related traits such as fasting glucose, body mass index, and fat distribution. These discoveries have largely been powered by the ability of researchers to undertake genome wide surveys for genetic associations in very large numbers of well-characterised samples, making use of high-density genotyping arrays capable of capturing the majority of common variation segregating in human populations [1].
- There are now over 40 loci confidently associated with individual risk of type 2 diabetes, and over 30 associated with body mass index and risk of obesity [2-4]. Each of these loci has the potential to reveal novel biological insights into disease pathogenesis, though a great deal of detailed functional work remains to be done to link the association signals discovered to the specific local transcripts through which they mediate their effect on disease risk.
- To date, most of these discoveries have been made in samples of European origin, whether collected in Europe or North America [2-4]. However, there are now growing numbers of genome wide association and resequencing studies for diabetes and related traits being conducted in samples from other parts of the world, most particularly those from East and South Asia, and from minority populations in the United States (Hispanics and African Americans) [5-10]. For example, recent studies of samples from East Asia were the first to describe type 2 diabetes risk variants near to the KCNQ1, UBE2E2, C2CD4A/B, SRR and PTPRD genes [5-8].
- This review will discuss the value of multiethnic studies of diabetes genetics, and describe how these are likely to add to our understanding of type 2 diabetes genetics and biology.
INTRODUCTION
- There have been a growing number of studies which have taken the diabetes association signals first discovered by genome wide association analyses in one population (mostly Europeans) and evaluated the evidence for their association with diabetes in others [11-16]. Moreover, as more and more genome wide association studies are completed in non-European populations [5-10], it becomes increasingly possible to compare the genome wide patterns of association across a wide diversity of ethnic groups.
- The consensus from these comparisons is that the majority of the signals identified so far show clear evidence of directionally-consistent association across major population groups [11-16]. This consistency is particularly obvious for samples from populations that are not of recent African origin: additional data from African-descent samples are awaited with interest. Initial reports of failed replication (at FTO for example) were largely, it seems, the result of inadequate sample size, combined with differences in allele frequency that made some signals far harder to detect in some non-European populations [17-21]. Given the small effect sizes of many of the common variant signals found so far, and the massive sample sizes required for their initial discovery, it is not surprising that most of these transethnic replication studies have been underpowered to detect confirmatory signals at all known loci. However, if one builds up data over multiple studies, and/or uses measures that are better-powered for modest sample size (such as genetic risk scores, or the proportion of loci showing directionally consistent odds ratios) the degree of overlap is striking [16]. Nor is this simply a case of loci that are discovered in Europeans being identified in other populations, as the reverse example of KCNQ1 demonstrates [5,6].
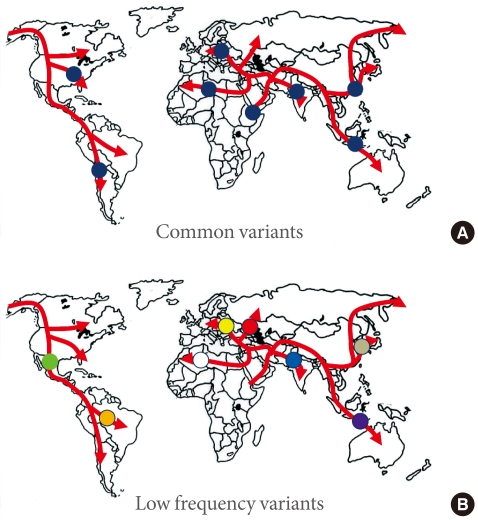
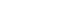
- What can we learn from this? The genome wide association studies conducted to date have necessarily focussed on common variants, and it should come as no surprise to find that most such variants are present in populations across the globe (Fig. 1). In the absence of strong selective pressures, it takes many thousands of generations for a mutation to drift to high frequency, and we can expect that most of the common variants seen in non-African populations predate the most recent expansion out of Africa, around 70,000 years ago, and will be shared amongst populations from Stockholm to Seoul, and from Mexico City to Mumbai [22].
- One obvious corollary of this overlap is that differences in the prevalence and in the presentation of diabetes across the world [23,24] are rather unlikely to be attributable to common sequence variants. Does this overlap mean that we should abandon efforts to map common variants for diabetes in additional populations, given the large investments already made in the analysis of samples from Europe and East Asia? Absolutely not, for the simple reason that the between-population differences in effect sizes and allele frequency that occur at some loci translate into very marked differences in the potential for their initial discovery (especially to the levels of statistical stringency required for genome wide studies). The loci emerging from genome wide association studies in East Asian samples demonstrate this extremely well: the signals at KCNQ1 and C2CD4A/B for example are definitely also present in European subjects but were missed by previous genome wide association efforts in Europeans for reasons of power and chance [5,6,8,25].
EXPLORING THE OVERLAP IN ASSOCIATION SIGNALS BETWEEN MAJOR ETHNIC GROUPS
- Genome wide association studies are reliant on linkage disequilibrium for the initial identification of signals since it is unlikely that the causal variant (or variants) at any locus will actually be represented on any given genotyping array. However, once a signal has been found and shown, by replication, to be genuine, linkage disequilibrium becomes an obstacle, frustrating efforts to home in on the causal variant at the locus. For example, at the FTO locus, attempts at further refinement of the association signal (through resequencing, dense genotyping or imputation from HapMap or 1,000 Genomes reference panels) have been unsuccessful: as far as we can tell, from studies of European samples at least, the causal allele could be any one of dozens of highly-correlated alleles carried on a 50 kb haplotype.
- However, since local patterns of linkage disequilibrium often differ between major population groups [22], one would hope that fine-mapping studies conducted at the transethnic level might enable some refinement of location, and in some circumstances, provide strong statistical evidence in favour of a single causal variant. Naturally there are some assumptions behind such analyses, the first being that the same single causal variant is shared between the populations concerned. The overlap in common variant signals reported above is clearly reassuring in this respect as it suggests that allelic heterogeneity is limited, at least amongst non-African populations.
- The major limitation of this approach is likely to be the fact that patterns of linkage disequilibrium and haplotype structure are quite similar between non-African populations [22], and this has fostered growing interest in the interrogation of samples of recent African origin (for example African Americans) [26]. The high genetic diversity of African populations, and the long period of divergence, means that the linkage disequilibrium patterns in African populations are often markedly different to those seen in Europeans and Asians. This has the potential therefore to offer considerable benefits in terms of fine-mapping, but only provided locus and allelic heterogeneity are not extreme. Put simply, there is a danger that at some loci, there will be no susceptibility alleles segregating in accessible African populations, meaning that there is "nothing to fine-map." The limited data for type 2 diabetes susceptibility in African Americans is reassuring in this respect [16,26], and it will be interesting to see the results of the genome wide association studies that are currently being completed using samples from this population.
- In the meantime, it seems sensible to pursue a broad strategy that attempts fine mapping in both non-African and African populations. Interestingly, several of the strongest diabetes susceptibility signals (TCF7L2, CDKAL1, and KCNQ1) do demonstrate rather unusually divergent haplotype structures between major ethnic groups [5,6,22], providing some encouragement that, as the data sets available become larger, effective fine-mapping will be possible. Fortunately such studies can be based around existing genome wide association data (complemented with imputation from ethnically-diverse reference panels, such as those forthcoming from the 1,000 Genomes Project [27]), so the costs are largely those of analysis.
TRANSETHNIC FINE MAPPING FOR COMMON VARIANT SIGNALS
- Transethnic studies are also capable of providing valuable insights into the genetic architecture of type 2 diabetes. An excellent example of this relates to the important clues that transethnic studies have provided with respect to the so-called "synthetic" association hypothesis [28]. This hypothesis, which was derived predominantly from simulation studies rather than empirical data, proposed that many (perhaps most) of the common variant signals identified by genome wide association studies are not the result of causal variants that are themselves also common. Rather, the common variant signals detected are merely a consequence of the ways in which multiple rare causal alleles at each locus are scattered across the common haplotypes in the region. If true, this "synthetic" association model has profound implications for the genetic architecture of common disease, and for the strategies that should be adopted for identification of the causal variants.
- Although widely promoted at the time of its publication, and seized upon by those antagonistic to the genome-wide association approach, there is relatively little empirical evidence to support this model. The CARD15 association with inflammatory bowel disease [29] shows that "synthetic" associations can occur, but are they really responsible for the majority of common variant signals detected by genome wide association studies?
- One clear prediction of the "synthetic" model is that common variant signals detected in one major ethnic group should not be expected to replicate in others. This is because rare alleles have usually arisen quite recently (in the absence of selection, it takes many generations for a new mutation to drift to higher frequency), such that many of these rare alleles will have appeared during the course of the modern human diaspora and will be not be widely-represented across multiple major ethnic groups (Fig. 1). Under those circumstances, it would be highly unlikely that the different sets of rare causal alleles that might have arisen in Europeans and East Asians (for example) would have stacked up, by chance, on the same set of haplotypes, and thereby generated the same common variant signals. However, this is precisely what appears what we observe for type 2 diabetes. In other words, the directional consistency and high reproducibility across major ethnic groups of almost all common variant signals for type 2 diabetes provides strong evidence that these signals are driven by causal alleles that are themselves common [16]. Presumably, these common, causal alleles predate the recent human expansion out of Africa, and having been carried to the four corners of the world, show broadly similar effects on diabetes risk.
- But can we go further? The thrifty genotype hypothesis, first promulgated by Neel, opined that the high prevalence of diabetes (and obesity) in modern populations, might be the result of many generations of selection for alleles that, in prehistoric times at least, conferred some kind of selective advantage [30]. The most obvious mechanism for this would involve individual differences in the capacity for the efficient storage of energy as fat during times of plenty. Individuals with "thrifty genotypes" would, according to this hypothesis, be in an advantageous position during periods of erratic food supply. However, in today's societies with access to constant (and excessive) food availability, individuals carrying these same alleles are now predisposed to develop obesity and diabetes.
- Given the growing number of diabetes-susceptibility variants now established, do transethnic comparisons provide evidence for selection that might support the thrifty genotype hypothesis? For the time being, the answer to this question remains far from conclusive. Using a variety of approaches, including comparing the frequencies of diabetes risk-alleles across populations, as well as looking for other genetic hallmarks of recent selection, studies to date have concluded that the evidence for selection is modest when viewed across all risk loci [31,32]. However, it is notable that the loci with the strongest evidence for ethnic differences in allele frequency and haplotype structure (both of these possible markers of selection) are also those (TCF7L2, CDKAL1, and KCNQ1) with some of the largest effects on diabetes risk [31,32]. It may be that the evidence for selection is most obvious when the phenotypic effects are also greatest and that these data are pointing towards subtle selection effects. Nonetheless, it is fair to say that the transethnic data to date fail to provide compelling support for the thrifty genotype hypothesis.
INFORMATION ON GENETIC ARCHITECTURE AND SELECTION
- Although most cases of diabetes across the globe are considered to fit within the "umbrella" of type 2 diabetes, there is no doubt that prevalence and presentation of type 2 diabetes differs between major ethnic groups [23,24,33,34]. Of course, these differences may turn out to be largely attributable to differences in environmental factors [35], but migration studies (for example the high prevalence of diabetes in migrant South Asian populations worldwide) may point to an important genetic contribution.
- For reasons hinted at above (particularly the high degree of overlap between the signals observed in different ethnic groups), it seems rather unlikely that between-population variation in the pattern of common variant signals will explain major differences in prevalence or presentation [16,33]. Having said that, there is emerging evidence that the effect sizes for most type 2 diabetes common variant loci are systematically larger in Japanese case-control comparisons than in equivalent analyses from other populations [16,36]. It remains to be seen whether this observation, if confirmed, represents an intrinsic and ethnic-specific different in genetic risk. Such differences in effect size could reflect the ways in which the cases and controls were selected (for example, selection for lean cases can boost some signals), the extent of environmental (dietary, economic) homogeneity (possibly greater in the Japanese population than in others), and the prevalence of obesity (if low, this may mean that cultural and lifestyle factors are having less of an impact on diabetes risk, thereby inflating the role of genetic variants).
- If there are genetic explanations for interethnic differences in prevalence and presentation of diabetes, these are likely to come from variants that are (at the global level at least) of lower frequency. Not only are such variants likely to be of more recent origin, and therefore more population specific (Fig. 1), but a subset of them may well have larger effects than the common variant signals discovered to date [37]. There are a growing number of examples of ethnic-specific variants that underlie substantial differences in disease prevalence - influencing rates of heart failure in South Asians, renal disease in Africans, and hepatosteatosis in Hispanics for example [38-40]. In some instances, these variants have been subject to marked selection and have risen to relatively high frequency in one or other major ethnic group. Whilst the detection of such highly-selected variants therefore continues to justify the application of common variant genome-wide association scan methodologies to diverse ethnic groups, it seems probable that resequencing approaches, directed towards low frequency and rare variant discovery, will prove the most powerful strategies for uncovering the genetic basis of interethnic differences in disease prevalence and presentation.
EXPLAINING DIFFERENCES IN PREVALENCE AND PRESENTATION OF DISEASE
- One of the major challenges thrown up by the success of the genome wide association approach lies in connecting the signals found to their downstream biology. Many of the genome wide association signals map to regulatory regions some distance from the nearest coding genes, and for only a minority of the forty or so known type 2 diabetes susceptibility loci has the transcript responsible for the causal effect been characterised [2]. This represents a serious impediment to the translation of the genetic discoveries into the improved understanding of disease predisposition that can support clinical advances.
- One of the most obvious strategies for linking signals to function lies in searching for "smoking gun" mutations in the genes mapping near to a genome wide association signal. The idea here is to expose the transcript responsible for the predisposition by identifying which (if any) of the genes in the vicinity contains variants predicted to have high functional impact (ideally rare, coding mutations of large effect that are clearly expected to abrogate gene function, such as frameshifts or premature stop mutations) and which can be shown to be responsible for type 2 diabetes or a closely related phenotype (such as a more genetic monogenic or syndromic form of diabetes). The best example of this approach to date comes from type 1 diabetes. Exon resequencing of the transcripts mapping to a genome wide association signal for type 1 diabetes on chromosome 2, revealed a number of low frequency variants with high putative functional impact within the IFIH1 gene, each of which showed evidence of an association with type 1 diabetes [41]. Though these variants did not explain the original common variant signal, they did provide a very strong pointer to IFIH1 as the gene most likely to be responsible for mediating the association effect at this locus.
- In conducting such studies, there are obvious merits in examining more than one ethnic group. Given that clear-cut "smoking gun" mutations (from both a statistical and functional perspective) will not be seen at every locus (they are likely to represent random accidents of nature, often of recent origin and likely to disappear within a few generations), extending the survey to a wide range of different ethnic groups provides the chance to "buy multiple tickets to the lottery." The hope is that an interesting "smoking gun" signal clearly visible in one ethnic group, can be rapidly followed up in others (where the signal exists but is not so obvious), and that it will be the accumulation of a wide variety of different "smoking gun" mutations, with clearly independent mutational histories, which provides the necessary pointers to identification of the transcript mediating the common variant association signal. The T2D-GENES consortium, for example, is testing this approach by resequencing over 500 genes from type 2 diabetes genome wide association signals in over 10,000 case-control samples ascertained from European, East Asian, South Asian, Hispanic, and African-American populations.
ADVANCING BIOLOGICAL UNDERSTANDING OF DISEASE PREDISPOSITION
- Human genetics is shifting from an era dominated by common variant discovery powered by genome-wide association studies, to one of low frequency and rare variant identification through sequencing. As the field moves in this direction, it will become ever more important, for a variety of reasons, to examine the genetic basis of disease in multiple ethnic groups. First, we can expect to see greater divergence of genetic predisposition between populations (both locus and allele heterogeneity) as far as low frequency variants are concerned, simply because they are more likely to be of recent, and ethnic-specific origin. Second, such divergence means that studies conducted in multiple ethnic groups (provided, of course, that they take proper account of population structure) will offer greater opportunities for discovery, and more chances to find high-impact alleles well-suited to subsequent functional and physiological characterisation. Third, as we've seen, the genetic basis of interethnic differences in prevalence and presentation of disease (including response to therapeutic and preventative interventions) is more likely to be explained by lower frequency variants. Fourth, as it becomes harder to obtain convincing statistical evidence within a single ethnic group that a given low frequency variant (or set of low frequency/rare variants) is associated with disease (simply because power for any given effect size is reduced for lower frequency variants), the demonstration that the same gene harbours an excess of rare susceptibility (or protective) alleles in several distinct ethnic groups will provide an ever more important signal for establishing a causal link between that gene and disease.
- For all these reasons, it is crucial, if we are to understand the basis of a global disease such as diabetes, that we pursue well-powered genetic and genomic enquiry in as many diverse populations as possible. It is equally important that these efforts are linked through strong scientific collaborations and mechanisms for data exchange, since it is increasingly true that, only by working together, will we be able to overcome the very considerable challenges that remain.
THE FUTURE
-
Acknowledgements
- I would like to acknowledge the many colleagues, senior and junior, national and international, with whom it has been such a pleasure to work on these challenging problems. The commitment to productive collaboration of researchers in our field has served as a powerful example to others. In particular, I wish to highlight the collective endeavour represented by the Global Diabetes Consortium and the T2D-GENES Consortium, funded by the National Institutes of Diabetes, Digestive and Kidney Diseases in the US (U01DK085545).
ACKNOWLEDGMENT
- 1. McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008;9:356-369. ArticlePubMedPDF
- 2. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Langenberg C, Hofmann OM, Dupuis J, Qi L, Segre AV, van Hoek M, Navarro P, Ardlie K, Balkau B, Benediktsson R, Bennett AJ, Blagieva R, Boerwinkle E, Bonnycastle LL, Bengtsson Bostrom K, Bravenboer B, Bumpstead S, Burtt NP, Charpentier G, Chines PS, Cornelis M, Couper DJ, Crawford G, Doney AS, Elliott KS, Elliott AL, Erdos MR, Fox CS, Franklin CS, Ganser M, Gieger C, Grarup N, Green T, Griffin S, Groves CJ, Guiducci C, Hadjadj S, Hassanali N, Herder C, Isomaa B, Jackson AU, Johnson PR, Jorgensen T, Kao WH, Klopp N, Kong A, Kraft P, Kuusisto J, Lauritzen T, Li M, Lieverse A, Lindgren CM, Lyssenko V, Marre M, Meitinger T, Midthjell K, Morken MA, Narisu N, Nilsson P, Owen KR, Payne F, Perry JR, Petersen AK, Platou C, Proenca C, Prokopenko I, Rathmann W, Rayner NW, Robertson NR, Rocheleau G, Roden M, Sampson MJ, Saxena R, Shields BM, Shrader P, Sigurdsson G, Sparso T, Strassburger K, Stringham HM, Sun Q, Swift AJ, Thorand B, Tichet J, Tuomi T, van Dam RM, van Haeften TW, van Herpt T, van Vliet-Ostaptchouk JV, Walters GB, Weedon MN, Wijmenga C, Witteman J, Bergman RN, Cauchi S, Collins FS, Gloyn AL, Gyllensten U, Hansen T, Hide WA, Hitman GA, Hofman A, Hunter DJ, Hveem K, Laakso M, Mohlke KL, Morris AD, Palmer CN, Pramstaller PP, Rudan I, Sijbrands E, Stein LD, Tuomilehto J, Uitterlinden A, Walker M, Wareham NJ, Watanabe RM, Abecasis GR, Boehm BO, Campbell H, Daly MJ, Hattersley AT, Hu FB, Meigs JB, Pankow JS, Pedersen O, Wichmann HE, Barroso I, Florez JC, Frayling TM, Groop L, Sladek R, Thorsteinsdottir U, Wilson JF, Illig T, Froguel P, van Duijn CM, Stefansson K, Altshuler D, Boehnke M, McCarthy MI. MAGIC investigators. GIANT Consortium. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010;42:579-589. ArticlePubMedPMCPDF
- 3. Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, Thorleifsson G, Zillikens MC, Speliotes EK, Magi R, Workalemahu T, White CC, Bouatia-Naji N, Harris TB, Berndt SI, Ingelsson E, Willer CJ, Weedon MN, Luan J, Vedantam S, Esko T, Kilpelainen TO, Kutalik Z, Li S, Monda KL, Dixon AL, Holmes CC, Kaplan LM, Liang L, Min JL, Moffatt MF, Molony C, Nicholson G, Schadt EE, Zondervan KT, Feitosa MF, Ferreira T, Allen HL, Weyant RJ, Wheeler E, Wood AR, Estrada K, Goddard ME, Lettre G, Mangino M, Nyholt DR, Purcell S, Smith AV, Visscher PM, Yang J, McCarroll SA, Nemesh J, Voight BF, Absher D, Amin N, Aspelund T, Coin L, Glazer NL, Hayward C, Heard-Costa NL, Hottenga JJ, Johansson A, Johnson T, Kaakinen M, Kapur K, Ketkar S, Knowles JW, Kraft P, Kraja AT, Lamina C, Leitzmann MF, McKnight B, Morris AP, Ong KK, Perry JR, Peters MJ, Polasek O, Prokopenko I, Rayner NW, Ripatti S, Rivadeneira F, Robertson NR, Sanna S, Sovio U, Surakka I, Teumer A, van Wingerden S, Vitart V, Zhao JH, Cavalcanti-Proenca C, Chines PS, Fisher E, Kulzer JR, Lecoeur C, Narisu N, Sandholt C, Scott LJ, Silander K, Stark K, Tammesoo ML, Teslovich TM, Timpson NJ, Watanabe RM, Welch R, Chasman DI, Cooper MN, Jansson JO, Kettunen J, Lawrence RW, Pellikka N, Perola M, Vandenput L, Alavere H, Almgren P, Atwood LD, Bennett AJ, Biffar R, Bonnycastle LL, Bornstein SR, Buchanan TA, Campbell H, Day IN, Dei M, Dorr M, Elliott P, Erdos MR, Eriksson JG, Freimer NB, Fu M, Gaget S, Geus EJ, Gjesing AP, Grallert H, Grassler J, Groves CJ, Guiducci C, Hartikainen AL, Hassanali N, Havulinna AS, Herzig KH, Hicks AA, Hui J, Igl W, Jousilahti P, Jula A, Kajantie E, Kinnunen L, Kolcic I, Koskinen S, Kovacs P, Kroemer HK, Krzelj V, Kuusisto J, Kvaloy K, Laitinen J, Lantieri O, Lathrop GM, Lokki ML, Luben RN, Ludwig B, McArdle WL, McCarthy A, Morken MA, Nelis M, Neville MJ, Pare G, Parker AN, Peden JF, Pichler I, Pietilainen KH, Platou CG, Pouta A, Ridderstrale M, Samani NJ, Saramies J, Sinisalo J, Smit JH, Strawbridge RJ, Stringham HM, Swift AJ, Teder-Laving M, Thomson B, Usala G, van Meurs JB, van Ommen GJ, Vatin V, Volpato CB, Wallaschofski H, Walters GB, Widen E, Wild SH, Willemsen G, Witte DR, Zgaga L, Zitting P, Beilby JP, James AL, Kahonen M, Lehtimaki T, Nieminen MS, Ohlsson C, Palmer LJ, Raitakari O, Ridker PM, Stumvoll M, Tonjes A, Viikari J, Balkau B, Ben-Shlomo Y, Bergman RN, Boeing H, Smith GD, Ebrahim S, Froguel P, Hansen T, Hengstenberg C, Hveem K, Isomaa B, Jorgensen T, Karpe F, Khaw KT, Laakso M, Lawlor DA, Marre M, Meitinger T, Metspalu A, Midthjell K, Pedersen O, Salomaa V, Schwarz PE, Tuomi T, Tuomilehto J, Valle TT, Wareham NJ, Arnold AM, Beckmann JS, Bergmann S, Boerwinkle E, Boomsma DI, Caulfield MJ, Collins FS, Eiriksdottir G, Gudnason V, Gyllensten U, Hamsten A, Hattersley AT, Hofman A, Hu FB, Illig T, Iribarren C, Jarvelin MR, Kao WH, Kaprio J, Launer LJ, Munroe PB, Oostra B, Penninx BW, Pramstaller PP, Psaty BM, Quertermous T, Rissanen A, Rudan I, Shuldiner AR, Soranzo N, Spector TD, Syvanen AC, Uda M, Uitterlinden A, Volzke H, Vollenweider P, Wilson JF, Witteman JC, Wright AF, Abecasis GR, Boehnke M, Borecki IB, Deloukas P, Frayling TM, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, North KE, O'Connell JR, Peltonen L, Schlessinger D, Strachan DP, Hirschhorn JN, Assimes TL, Wichmann HE, Thorsteinsdottir U, van Duijn CM, Stefansson K, Cupples LA, Loos RJ, Barroso I, McCarthy MI, Fox CS, Mohlke KL, Lindgren CM. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet 2010;42:949-960. ArticlePubMedPMCPDF
- 4. Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Allen HL, Lindgren CM, Luan J, Magi R, Randall JC, Vedantam S, Winkler TW, Qi L, Workalemahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant RJ, Segre AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpelainen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AL, Fisher E, Glazer NL, Goddard ME, Heard-Costa NL, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JR, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MT, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AI, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, Busonero F, Campbell H, Cappuccio FP, Cavalcanti-Proenca C, Chen YD, Chen CM, Chines PS, Clarke R, Coin L, Connell J, Day IN, Heijer M, Duan J, Ebrahim S, Elliott P, Elosua R, Eiriksdottir G, Erdos MR, Eriksson JG, Facheris MF, Felix SB, Fischer-Posovszky P, Folsom AR, Friedrich N, Freimer NB, Fu M, Gaget S, Gejman PV, Geus EJ, Gieger C, Gjesing AP, Goel A, Goyette P, Grallert H, Grassler J, Greenawalt DM, Groves CJ, Gudnason V, Guiducci C, Hartikainen AL, Hassanali N, Hall AS, Havulinna AS, Hayward C, Heath AC, Hengstenberg C, Hicks AA, Hinney A, Hofman A, Homuth G, Hui J, Igl W, Iribarren C, Isomaa B, Jacobs KB, Jarick I, Jewell E, John U, Jorgensen T, Jousilahti P, Jula A, Kaakinen M, Kajantie E, Kaplan LM, Kathiresan S, Kettunen J, Kinnunen L, Knowles JW, Kolcic I, Konig IR, Koskinen S, Kovacs P, Kuusisto J, Kraft P, Kvaloy K, Laitinen J, Lantieri O, Lanzani C, Launer LJ, Lecoeur C, Lehtimaki T, Lettre G, Liu J, Lokki ML, Lorentzon M, Luben RN, Ludwig B, Manunta P, Marek D, Marre M, Martin NG, McArdle WL, McCarthy A, McKnight B, Meitinger T, Melander O, Meyre D, Midthjell K, Montgomery GW, Morken MA, Morris AP, Mulic R, Ngwa JS, Nelis M, Neville MJ, Nyholt DR, O'Donnell CJ, O'Rahilly S, Ong KK, Oostra B, Pare G, Parker AN, Perola M, Pichler I, Pietilainen KH, Platou CG, Polasek O, Pouta A, Rafelt S, Raitakari O, Rayner NW, Ridderstrale M, Rief W, Ruokonen A, Robertson NR, Rzehak P, Salomaa V, Sanders AR, Sandhu MS, Sanna S, Saramies J, Savolainen MJ, Scherag S, Schipf S, Schreiber S, Schunkert H, Silander K, Sinisalo J, Siscovick DS, Smit JH, Soranzo N, Sovio U, Stephens J, Surakka I, Swift AJ, Tammesoo ML, Tardif JC, Teder-Laving M, Teslovich TM, Thompson JR, Thomson B, Tonjes A, Tuomi T, van Meurs JB, van Ommen GJ, Vatin V, Viikari J, Visvikis-Siest S, Vitart V, Vogel CI, Voight BF, Waite LL, Wallaschofski H, Walters GB, Widen E, Wiegand S, Wild SH, Willemsen G, Witte DR, Witteman JC, Xu J, Zhang Q, Zgaga L, Ziegler A, Zitting P, Beilby JP, Farooqi IS, Hebebrand J, Huikuri HV, James AL, Kahonen M, Levinson DF, Macciardi F, Nieminen MS, Ohlsson C, Palmer LJ, Ridker PM, Stumvoll M, Beckmann JS, Boeing H, Boerwinkle E, Boomsma DI, Caulfield MJ, Chanock SJ, Collins FS, Cupples LA, Smith GD, Erdmann J, Froguel P, Gronberg H, Gyllensten U, Hall P, Hansen T, Harris TB, Hattersley AT, Hayes RB, Heinrich J, Hu FB, Hveem K, Illig T, Jarvelin MR, Kaprio J, Karpe F, Khaw KT, Kiemeney LA, Krude H, Laakso M, Lawlor DA, Metspalu A, Munroe PB, Ouwehand WH, Pedersen O, Penninx BW, Peters A, Pramstaller PP, Quertermous T, Reinehr T, Rissanen A, Rudan I, Samani NJ, Schwarz PE, Shuldiner AR, Spector TD, Tuomilehto J, Uda M, Uitterlinden A, Valle TT, Wabitsch M, Waeber G, Wareham NJ, Watkins H, Wilson JF, Wright AF, Zillikens MC, Chatterjee N, McCarroll SA, Purcell S, Schadt EE, Visscher PM, Assimes TL, Borecki IB, Deloukas P, Fox CS, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, Mohlke KL, O'Connell JR, Peltonen L, Schlessinger D, Strachan DP, van Duijn CM, Wichmann HE, Frayling TM, Thorsteinsdottir U, Abecasis GR, Barroso I, Boehnke M, Stefansson K, North KE, McCarthy MI, Hirschhorn JN, Ingelsson E, Loos RJ. MAGIC. Procardis Consortium. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010;42:937-948. ArticlePubMedPMCPDF
- 5. Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A, Sato Y, Yamagata K, Hinokio Y, Wang HY, Tanahashi T, Nakamura N, Oka Y, Iwasaki N, Iwamoto Y, Yamada Y, Seino Y, Maegawa H, Kashiwagi A, Takeda J, Maeda E, Shin HD, Cho YM, Park KS, Lee HK, Ng MC, Ma RC, So WY, Chan JC, Lyssenko V, Tuomi T, Nilsson P, Groop L, Kamatani N, Sekine A, Nakamura Y, Yamamoto K, Yoshida T, Tokunaga K, Itakura M, Makino H, Nanjo K, Kadowaki T, Kasuga M. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008;40:1092-1097. ArticlePubMedPDF
- 6. Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Ng DP, Holmkvist J, Borch-Johnsen K, Jorgensen T, Sandbaek A, Lauritzen T, Hansen T, Nurbaya S, Tsunoda T, Kubo M, Babazono T, Hirose H, Hayashi M, Iwamoto Y, Kashiwagi A, Kaku K, Kawamori R, Tai ES, Pedersen O, Kamatani N, Kadowaki T, Kikkawa R, Nakamura Y, Maeda S. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 2008;40:1098-1102. ArticlePubMedPDF
- 7. Tsai FJ, Yang CF, Chen CC, Chuang LM, Lu CH, Chang CT, Wang TY, Chen RH, Shiu CF, Liu YM, Chang CC, Chen P, Chen CH, Fann CS, Chen YT, Wu JY. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet 2010;6:e1000847ArticlePubMedPMC
- 8. Yamauchi T, Hara K, Maeda S, Yasuda K, Takahashi A, Horikoshi M, Nakamura M, Fujita H, Grarup N, Cauchi S, Ng DP, Ma RC, Tsunoda T, Kubo M, Watada H, Maegawa H, Okada-Iwabu M, Iwabu M, Shojima N, Shin HD, Andersen G, Witte DR, Jorgensen T, Lauritzen T, Sandbæk A, Hansen T, Ohshige T, Omori S, Saito I, Kaku K, Hirose H, So WY, Beury D, Chan JC, Park KS, Tai ES, Ito C, Tanaka Y, Kashiwagi A, Kawamori R, Kasuga M, Froguel P, Pedersen O, Kamatani N, Nakamura Y, Kadowaki T. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat Genet 2010;42:864-868. ArticlePubMedPDF
- 9. Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban HJ, Yoon D, Lee MH, Kim DJ, Park M, Cha SH, Kim JW, Han BG, Min H, Ahn Y, Park MS, Han HR, Jang HY, Cho EY, Lee JE, Cho NH, Shin C, Park T, Park JW, Lee JK, Cardon L, Clarke G, McCarthy MI, Lee JY, Lee JK, Oh B, Kim HL. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet 2009;41:527-534. ArticlePubMedPDF
- 10. Chambers JC, Zhang W, Zabaneh D, Sehmi J, Jain P, McCarthy MI, Froguel P, Ruokonen A, Balding D, Jarvelin MR, Scott J, Elliott P, Kooner JS. Common genetic variation near melatonin receptor MTNR1B contributes to raised plasma glucose and increased risk of type 2 diabetes among Indian Asians and European Caucasians. Diabetes 2009;58:2703-2708. ArticlePubMedPMCPDF
- 11. Sanghera DK, Ortega L, Han S, Singh J, Ralhan SK, Wander GS, Mehra NK, Mulvihill JJ, Ferrell RE, Nath SK, Kamboh MI. Impact of nine common type 2 diabetes risk polymorphisms in Asian Indian Sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTO variants confer a significant risk. BMC Med Genet 2008;9:59ArticlePubMedPMCPDF
- 12. Horikawa Y, Miyake K, Yasuda K, Enya M, Hirota Y, Yamagata K, Hinokio Y, Oka Y, Iwasaki N, Iwamoto Y, Yamada Y, Seino Y, Maegawa H, Kashiwagi A, Yamamoto K, Tokunaga K, Takeda J, Kasuga M. Replication of genome-wide association studies of type 2 diabetes susceptibility in Japan. J Clin Endocrinol Metab 2008;93:3136-3141. ArticlePubMedPDF
- 13. Lee YH, Kang ES, Kim SH, Han SJ, Kim CH, Kim HJ, Ahn CW, Cha BS, Nam M, Nam CM, Lee HC. Association between polymorphisms in SLC30A8, HHEX, CDKN2A/B, IGF2BP2, FTO, WFS1, CDKAL1, KCNQ1 and type 2 diabetes in the Korean population. J Hum Genet 2008;53:991-998. ArticlePubMedPDF
- 14. Ng MC, Park KS, Oh B, Tam CH, Cho YM, Shin HD, Lam VK, Ma RC, So WY, Cho YS, Kim HL, Lee HK, Chan JC, Cho NH. Implication of genetic variants near TCF7L2, SLC30A8, HHEX, CDKAL1, CDKN2A/B, IGF2BP2, and FTO in type 2 diabetes and obesity in 6,719 Asians. Diabetes 2008;57:2226-2233. ArticlePubMedPMCPDF
- 15. Lewis JP, Palmer ND, Hicks PJ, Sale MM, Langefeld CD, Freedman BI, Divers J, Bowden DW. Association analysis in African Americans of European-derived type 2 diabetes single nucleotide polymorphisms from whole-genome association studies. Diabetes 2008;57:2220-2225. ArticlePubMedPMCPDF
- 16. Waters KM, Stram DO, Hassanein MT, Le Marchand L, Wilkens LR, Maskarinec G, Monroe KR, Kolonel LN, Altshuler D, Henderson BE, Haiman CA. Consistent association of type 2 diabetes risk variants found in europeans in diverse racial and ethnic groups. PLoS Genet 2010;6(8):pii:e1001078.Article
- 17. Li H, Wu Y, Loos RJ, Hu FB, Liu Y, Wang J, Yu Z, Lin X. Variants in the fat mass- and obesity-associated (FTO) gene are not associated with obesity in a Chinese Han population. Diabetes 2008;57:264-268. ArticlePubMedPDF
- 18. Radha V, Vimaleswaran KS, Babu HN, Abate N, Chandalia M, Satija P, Grundy SM, Ghosh S, Majumder PP, Deepa R, Rao SM, Mohan V. Role of genetic polymorphism peroxisome proliferator-activated receptor-gamma2 Pro12Ala on ethnic susceptibility to diabetes in South-Asian and Caucasian subjects: evidence for heterogeneity. Diabetes Care 2006;29:1046-1051. ArticlePubMed
- 19. Chang YC, Liu PH, Lee WJ, Chang TJ, Jiang YD, Li HY, Kuo SS, Lee KC, Chuang LM. Common variation in the fat mass and obesity-associated (FTO) gene confers risk of obesity and modulates BMI in the Chinese population. Diabetes 2008;57:2245-2252. ArticlePubMedPMCPDF
- 20. Tan JT, Dorajoo R, Seielstad M, Sim XL, Ong RT, Chia KS, Wong TY, Saw SM, Chew SK, Aung T, Tai ES. FTO variants are associated with obesity in the Chinese and Malay populations in Singapore. Diabetes 2008;57:2851-2857. ArticlePubMedPMCPDF
- 21. Chandak GR, Janipalli CS, Bhaskar S, Kulkarni SR, Mohankrishna P, Hattersley AT, Frayling TM, Yajnik CS. Common variants in the TCF7L2 gene are strongly associated with type 2 diabetes mellitus in the Indian population. Diabetologia 2007;50:63-67. ArticlePubMedPDF
- 22. International HapMap 3 Consortium. Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Peltonen L, Dermitzakis E, Bonnen PE, Altshuler DM, Gibbs RA, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Yu F, Chang K, Hawes A, Lewis LR, Ren Y, Wheeler D, Gibbs RA, Muzny DM, Barnes C, Darvishi K, Hurles M, Korn JM, Kristiansson K, Lee C, McCarrol SA, Nemesh J, Dermitzakis E, Keinan A, Montgomery SB, Pollack S, Price AL, Soranzo N, Bonnen PE, Gibbs RA, Gonzaga-Jauregui C, Keinan A, Price AL, Yu F, Anttila V, Brodeur W, Daly MJ, Leslie S, McVean G, Moutsianas L, Nguyen H, Schaffner SF, Zhang Q, Ghori MJ, McGinnis R, McLaren W, Pollack S, Price AL, Schaffner SF, Takeuchi F, Grossman SR, Shlyakhter I, Hostetter EB, Sabeti PC, Adebamowo CA, Foster MW, Gordon DR, Licinio J, Manca MC, Marshall PA, Matsuda I, Ngare D, Wang VO, Reddy D, Rotimi CN, Royal CD, Sharp RR, Zeng C, Brooks LD, McEwen JE. Integrating common and rare genetic variation in diverse human populations. Nature 2010;467:52-58. ArticlePubMedPMCPDF
- 23. Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001;414:782-787. ArticlePubMedPDF
- 24. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004;27:1047-1053. ArticlePubMedPDF
- 25. McCarthy MI. Casting a wider net for diabetes susceptibility genes. Nat Genet 2008;40:1039-1040. ArticlePubMedPDF
- 26. Hassanein MT, Lyon HN, Nguyen TT, Akylbekova EL, Waters K, Lettre G, Tayo B, Forrester T, Sarpong DF, Stram DO, Butler JL, Wilks R, Liu J, Le Marchand L, Kolonel LN, Zhu X, Henderson B, Cooper R, McKenzie C, Taylor HA Jr, Haiman CA, Hirschhorn JN. Fine mapping of the association with obesity at the FTO locus in African-derived populations. Hum Mol Genet 2010;19:2907-2916. ArticlePubMedPMC
- 27. 1000 Genomes Project Consortium. Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061-1073. ArticlePubMedPMCPDF
- 28. Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol 2010;8:e1000294ArticlePubMedPMC
- 29. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001;411:599-603. ArticlePubMedPDF
- 30. NEEL JV. Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress"? Am J Hum Genet 1962;14:353-362. PubMedPMC
- 31. Southam L, Soranzo N, Montgomery SB, Frayling TM, McCarthy MI, Barroso I, Zeggini E. Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes-and obesity-susceptibility variants? Diabetologia 2009;52:1846-1851. ArticlePubMedPMC
- 32. Helgason A, Palsson S, Thorleifsson G, Grant SF, Emilsson V, Gunnarsdottir S, Adeyemo A, Chen Y, Chen G, Reynisdottir I, Benediktsson R, Hinney A, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Schafer H, Faruque M, Doumatey A, Zhou J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Sigurdsson G, Hebebrand J, Pedersen O, Thorsteinsdottir U, Gulcher JR, Kong A, Rotimi C, Stefansson K. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet 2007;39:218-225. ArticlePubMedPDF
- 33. Takeuchi M, Okamoto K, Takagi T, Ishii H. Ethnic difference in patients with type 2 diabetes mellitus in inter-East Asian populations: a systematic review and meta-analysis focusing on gene polymorphism. J Diabetes 2009;1:255-262. ArticlePubMedPDF
- 34. McKeigue PM, Shah B, Marmot MG. Relation of central obesity and insulin resistance with high diabetes prevalence and cardiovascular risk in South Asians. Lancet 1991;337:382-386. ArticlePubMed
- 35. Florez JC, Price AL, Campbell D, Riba L, Parra MV, Yu F, Duque C, Saxena R, Gallego N, Tello-Ruiz M, Franco L, Rodriguez-Torres M, Villegas A, Bedoya G, Aguilar-Salinas CA, Tusie-Luna MT, Ruiz-Linares A, Reich D. Strong association of socioeconomic status with genetic ancestry in Latinos: implications for admixture studies of type 2 diabetes. Diabetologia 2009;52:1528-1536. ArticlePubMedPMCPDF
- 36. Takeuchi F, Serizawa M, Yamamoto K, Fujisawa T, Nakashima E, Ohnaka K, Ikegami H, Sugiyama T, Katsuya T, Miyagishi M, Nakashima N, Nawata H, Nakamura J, Kono S, Takayanagi R, Kato N. Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes 2009;58:1690-1699. ArticlePubMedPMCPDF
- 37. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature 2009;461:747-753. ArticlePubMedPMCPDF
- 38. Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, Tharkan JM, Vaideeswar P, Rathinavel A, Narasimhan C, Ayapati DR, Ayub Q, Mehdi SQ, Oppenheimer S, Richards MB, Price AL, Patterson N, Reich D, Singh L, Tyler-Smith C, Thangaraj K. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet 2009;41:187-191. ArticlePubMedPMCPDF
- 39. Genovese G, Tonna SJ, Knob AU, Appel GB, Katz A, Bernhardy AJ, Needham AW, Lazarus R, Pollak MR. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int 2010;78:698-704. ArticlePubMedPMC
- 40. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461-1465. ArticlePubMedPMCPDF
- 41. Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009;324:387-389. ArticlePubMedPMC
REFERENCES
Fig. 1Common variants (A) will typically have arisen prior to the modern human exodus from Africa, and will be widely represented amongst non-African populations. In contrast, lower-frequency alleles (B) will usually be of more recent origin, and to have arisen during the diaspora: such alleles will not be widely-represented, and may be restricted to a particular population.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Validation and genetic heritability estimation of known type 2 diabetes related variants in the Korean population
Hye-Mi Jang, Mi Yeong Hwang, Bong-Jo Kim, Young Jin Kim
Genomics & Informatics.2021; 19(4): e37. CrossRef - Distinct subtypes of polycystic ovary syndrome with novel genetic associations: An unsupervised, phenotypic clustering analysis
Matthew Dapas, Frederick T. J. Lin, Girish N. Nadkarni, Ryan Sisk, Richard S. Legro, Margrit Urbanek, M. Geoffrey Hayes, Andrea Dunaif, Jenny E. Myers
PLOS Medicine.2020; 17(6): e1003132. CrossRef - Association of a genetic variant of the ZPR1 zinc finger gene with type 2 diabetes mellitus
FUMITAKA TOKORO, REIKO MATSUOKA, SHINTARO ABE, MASAZUMI ARAI, TOSHIYUKI NODA, SACHIRO WATANABE, HIDEKI HORIBE, TETSUO FUJIMAKI, MITSUTOSHI OGURI, KIMIHIKO KATO, SHINYA MINATOGUCHI, YOSHIJI YAMADA
Biomedical Reports.2015; 3(1): 88. CrossRef - Insights into the Genetic Susceptibility to Type 2 Diabetes from Genome-Wide Association Studies of Glycaemic Traits
Letizia Marullo, Julia S. El-Sayed Moustafa, Inga Prokopenko
Current Diabetes Reports.2014;[Epub] CrossRef - Towards Virtual Knowledge Broker services for semantic integration of life science literature and data sources
Ian Harrow, Wendy Filsell, Peter Woollard, Ian Dix, Michael Braxenthaler, Richard Gedye, David Hoole, Richard Kidd, Jabe Wilson, Dietrich Rebholz-Schuhmann
Drug Discovery Today.2013; 18(9-10): 428. CrossRef - Genome-Wide Association Study for Type 2 Diabetes in Indians Identifies a New Susceptibility Locus at 2q21
Rubina Tabassum, Ganesh Chauhan, Om Prakash Dwivedi, Anubha Mahajan, Alok Jaiswal, Ismeet Kaur, Khushdeep Bandesh, Tejbir Singh, Benan John Mathai, Yogesh Pandey, Manickam Chidambaram, Amitabh Sharma, Sreenivas Chavali, Shantanu Sengupta, Lakshmi Ramakris
Diabetes.2013; 62(3): 977. CrossRef - A replication study of 19 GWAS-validated type 2 diabetes at-risk variants in the Lebanese population
Wassim Y. Almawi, Rita Nemr, Sose H. Keleshian, Akram Echtay, Fabiola Lisa Saldanha, Fatima A. AlDoseri, Eddie Racoubian
Diabetes Research and Clinical Practice.2013; 102(2): 117. CrossRef - Single nucleotide polymorphisms in JAZF1 and BCL11A gene are nominally associated with type 2 diabetes in African-American families from the GENNID study
Kurt A Langberg, Lijun Ma, Neeraj K Sharma, Craig L Hanis, Steven C Elbein, Sandra J Hasstedt, Swapan K Das
Journal of Human Genetics.2012; 57(1): 57. CrossRef - Typ-2-Diabetes-assoziierte Gene
J. Kriebel, H. Grallert, T. Illig
Der Diabetologe.2012; 8(1): 26. CrossRef - Genomweite Assoziationsstudien (GWAS) — Möglichkeiten und Grenzen
Jennifer Kriebel, Thomas Illig, Harald Grallert
BIOspektrum.2012; 18(5): 508. CrossRef - T2DM: Why Epigenetics?
Delphine Fradin, Pierre Bougnères
Journal of Nutrition and Metabolism.2011; 2011: 1. CrossRef
 PubReader
PubReader Cite
Cite