- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Ahead-of print > Article
-
ReviewPathophysiology Dysfunctional Mitochondria Clearance in Situ: Mitophagy in Obesity and Diabetes-Associated Cardiometabolic Diseases
-
Songling Tang1*
 , Di Hao1*, Wen Ma2*, Lian Liu1, Jiuyu Gao1, Peng Yao1, Haifang Yu1, Lu Gan1, Yu Cao1,3
, Di Hao1*, Wen Ma2*, Lian Liu1, Jiuyu Gao1, Peng Yao1, Haifang Yu1, Lu Gan1, Yu Cao1,3 -
DOI: https://doi.org/10.4093/dmj.2023.0213
Published online: February 15, 2024
- 1,134 Views
- 67 Download
1Department of Emergency Medicine, Laboratory of Emergency Medicine, West China Hospital, West China School of Medicine, Sichuan University, Chengdu, China
2Sichuan University-The Hong Kong Polytechnic University Institute for Disaster Management and Reconstruction, Chengdu, China
3Disaster Medical Center, Sichuan University, Chengdu, China
-
Corresponding authors: Lu Gan Department of Emergency Medicine, Laboratory of Emergency Medicine, West China Hospital, West China School of Medicine, Sichuan University, 37 Guoxue Road, Chengdu 610041, Sichuan, China E-mail: ganlu@wchscu.cn
-
Yu Cao Department of Emergency Medicine, Laboratory of Emergency Medicine, West China Hospital, West China School of Medicine, Sichuan University, 37 Guoxue Road, Chengdu 610041, Sichuan, China E-mail: caoyu@wchscu.cn
- *Songling Tang, Di Hao, and Wen Ma contributed equally to this study as first authors.
Copyright © 2024 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- KEY FIGURE
- Highlights
- INTRODUCTION
- MITOCHONDRIAL DYSFUNCTION IS AN IMPORTANT PATHOLOGICAL MANIFESTATION OF OBESITY AND DIABETES-RELATED HEART DISORDERS
- MITOPHAGY IS REQUIRED TO CLEAR DYSFUNCTIONAL MITOCHONDRIAL
- SUPPRESSED AND EXCESSIVE MITOPHAGY ARE BOTH OBSERVED IN OBESITY AND DIABETES-RELATED HEART DISORDERS
- REGULATION OF THE MITOPHAGY PATHWAY IN OBESITY AND DIABETES-RELATED HEART DISORDERS
- PHARMACOLOGIC INTERVENTION IN MITOPHAGY IN OBESITY AND DIABETES-RELATED HEART DISORDERS
- CONCLUSIONS
- NOTES
- REFERENCES
ABSTRACT
- Several mitochondrial dysfunctions in obesity and diabetes include impaired mitochondrial membrane potential, excessive mitochondrial reactive oxygen species generation, reduced mitochondrial DNA, increased mitochondrial Ca2+ flux, and mitochondrial dynamics disorders. Mitophagy, specialized autophagy, is responsible for clearing dysfunctional mitochondria in physiological and pathological conditions. As a paradox, inhibition and activation of mitophagy have been observed in obesity and diabetes-related heart disorders, with both exerting bidirectional effects. Suppressed mitophagy is beneficial to mitochondrial homeostasis, also known as benign mitophagy. On the contrary, in most cases, excessive mitophagy is harmful to dysfunctional mitochondria elimination and thus is defined as detrimental mitophagy. In obesity and diabetes, two classical pathways appear to regulate mitophagy, including PTEN-induced putative kinase 1 (PINK1)/Parkin-dependent mitophagy and receptors/adapters-dependent mitophagy. After the pharmacologic interventions of mitophagy, mitochondrial morphology and function have been restored, and cell viability has been further improved. Herein, we summarize the mitochondrial dysfunction and mitophagy alterations in obesity and diabetes, as well as the underlying upstream mechanisms, in order to provide novel therapeutic strategies for the obesity and diabetes-related heart disorders.
- • Mitochondrial dysfunction is a crucial pathology in obesity and diabetes.
- • Mitochondria is responsible for the selective removal of impaired mitochondria.
- • Mitophagy dysfunction varies with tissue and time, seen in both suppressed and activated states.
- • Interventions targeting mitophagy could be beneficial for managing obesity and diabetes.
Highlights
- As well-acknowledged, autophagy is crucial for clearing abnormal cells and cellular self-protection. Dysfunctional organelles and some specific foreign bodies can be cleared by autophagy, which is defined as selective autophagy. There are particular autophagy pathways in mammals, such as mitophagy, proteaphagy, ribophagy, pexophagy, endoplasmic reticulum (ER)- phagy, lysophagy, lipophagy, and nucleophagy [1]. Additionally, selective autophagy is observed explicitly in yeast, such as xenophagy for pathogen clearance [2]. Mitophagy is responsible for the selective removal of impaired mitochondria, and it is considered a cellular self-protective process under both physiological and pathological conditions [3]. The obesity and diabetes-related heart disorders refer to the common metabolic-related structural impairments and cardiac dysfunctions caused by cell senescence, autophagy and apoptosis in obesity and diabetes, which is different from the macrovascular complications of obesity and diabetes [4]. Among them, diabetes-induced cardiomyopathy, which is also called diabetic cardiomyopathy, is a typical phenotype in obesity and diabetes-related heart disorders and is associated with significant mitochondrial dysfunction, ER stress, and impaired calcium homeostasis [5]. Under pathological conditions in obesity and diabetes-related heart disorders, due to significant mitochondrial dysfunction, paradoxically abnormal mitophagy occurs [6], including defective but beneficial mitophagy and excessive but detrimental mitophagy [7,8]. According to the different alterations in mitophagy, pharmacologic or genetic interventions, including suppression and activation, can be performed, and remarkable protective effects have been revealed. However, different tissues and phases of the disease have shown significant differences, and the mechanisms are not fully understood. Therefore, we will summarize the alterations in mitophagy in obesity and diabetes-related heart disorders, as well as the effects and underlying mechanisms, to investigate potential therapeutic targets.
INTRODUCTION
- In physiological conditions, mitochondria are responsible for adenosine triphosphate (ATP) generation [9], metabolic homeostasis maintenance, redox homeostasis [10], calcium regulation, cell survival, and cell death [11]. Once mitochondria are damaged, mitochondrial dysfunction occurs, such as mitochondrial dynamics disorders (mitochondrial fission or fusion cycling disorders) [12,13], excessive mitochondrial reactive oxygen species (mtROS) production [14], and abnormal mitochondrial DNA (mtDNA) accumulation [12]. As a result, the disruption of mitochondrial function contributes to cellular dysfunction and cell death.
- Obesity and diabetes are well-acknowledged energy metabolism disorders, and multiple mitochondrial impairments have been observed in obesity and diabetes-related heart disorders, such as mitochondrial deformation, impaired mitochondrial potential, excessive generation of mtROS, reduced mtDNA levels, increased mitochondrial Ca2+ flux, and disturbances in mitochondrial fission and fusion. First, increases in mitochondrial number and area, indicating abnormal mitochondrial accumulation, have been detected in mouse models of high-fat diet (HFD)-induced obesity and diabetes [6]. Second, swollen mitochondria and enlarged mitochondrial volume, which are types of mitochondrial deformation, are observed in mice with diabetes [15]. Consistent with this finding, impaired mitochondrial respiratory function is found in obese mice due to decreased expression of the mitochondrial respiration complex I, III, and IV, resulting in reduced ATP production [16,17]. Third, obesity and diabetes have been reported to decrease mitochondrial membrane potential, as measured by JC-1 staining, in HFD-induced in vivo models and palmitic acid (PA)- induced in vitro models, which indicates pathological mitochondrial membrane dysfunction and increased mitochondrial permeability in obesity [18,19]. Fourth, due to chronic inflammatory stimulation in individuals with obesity and diabetes, excessive mtROS, including superoxide (O2–) and hydrogen peroxide (H2O2), are generated, mtDNA copy number is decreased, and there is a consequent decline in the mitochondrial DNA to nuclear DNA ratio (mtDNA:nDNA) [17,20]. Finally, altered mitochondrial dynamics have been reported in response to high-fat (HF)-induced stress, resulting in significant upregulation of the mitochondrial fission protein dynamin related protein 1 (Drp1) and downregulation of the mitochondrial fusion proteins mitofusin 2 (Mfn2) and mitochondrial dynamin like GTPase (OPA1) [21-23]. Overall, these mitochondrial disorders lead to dysfunctional mitochondrial accumulation, a reduction in ATP production, mitochondrial disruption, cellular degeneration, and cell death, such as apoptosis.
MITOCHONDRIAL DYSFUNCTION IS AN IMPORTANT PATHOLOGICAL MANIFESTATION OF OBESITY AND DIABETES-RELATED HEART DISORDERS
- Mitophagy is crucial for maintaining mitochondrial quantity, mitochondrial quality, and mitochondrial metabolic homeostasis. Physiological and pathological mitophagy lead to mitochondrial clearance. Under certain physiological conditions, mitophagy can be induced by aged and/or damaged mitochondrial degeneration. Physiological mitophagy can be classified as basal mitophagy, stress-induced mitophagy and programmed mitophagy [24]. Studies on basal mitophagy, which is also known as steady-state mitophagy, are limited. McWilliams et al. and Sun et al.’s studies [25,26] revealed basal mitophagy under physiological conditions with cell-type and tissue-specific differences. In response to external stress, such as hypoxia [27], stress-induced mitophagy is induced, which mediates metabolic homeostasis through mitochondrial quality control [24]. Mitochondrial clearance during the development of several specific cell types is defined as programmed mitophagy. Mitochondria are eliminated from the cytoplasm during the maturation of erythrocytes [28], and this process is called programmed mitophagy. The elimination of paternal mitochondria during the development of fertilized oocytes [29] is also due to programmed mitophagy, and this process is regulated by the RAB7 member of the RAS oncogene family, which is a newly discovered factor [30].
- Pathological mitophagy [24] often occurs during aging and in other pathological conditions, such as metabolic disorders such as HFD-induced obesity [31], cardiovascular diseases [12], neurodegenerative diseases such as Alzheimer’s disease (AD) [32], and inflammation such as sepsis [33]. Emerging evidence suggests that mitophagy contributes to the onset and progression of major neurodegenerative diseases. Defective mitophagy is found in AD patients and animal models, and impaired removal of dysfunctional mitochondria through mitophagy is observed in Parkinson’s disease and Huntington’s disease [34]. In lipopolysaccharide-induced septic animal models, mitophagy is inhibited in activated macrophages, thus contributing to the survival of activated macrophages [33]. In addition, impaired autophagic flux and mitophagy have been observed in Lactobacillus casei cell wall extract-induced Kawasaki disease murine models, along with the excessive accumulation of mtROS and mtDNA in cardiovascular lesions due to the pathological decline in dysfunctional mitochondrial clearance [35].
MITOPHAGY IS REQUIRED TO CLEAR DYSFUNCTIONAL MITOCHONDRIAL
- To date, it remains unclear how mitophagy is altered in obese individuals and type 2 diabetes mellitus (T2DM) patients. However, many researchers have explored how mitophagy changes and the possible mechanisms. In clinical patients, defective mitophagy is observed in obesity and diabetes, and suppressed expression of mitophagy markers, such as microtubule-associated protein 1 light chain 3 beta (LC3B), PTEN-induced putative kinase 1 (PINK1), and Parkin, has been observed [36,37]. However, the changes in mitophagy changes under high glucose (HG) or HF stress in obese and diabetic animal models and in vitro are controversial because activated mitophagy and repressed mitophagy have been observed in various studies. The difference in mitophagy might be time-dependent, since mitophagy status could be different in the early and later phases of HFD consumption. In the cardiomyocytes of HFD-fed mice [38], autophagy is altered in a time-dependent manner and is upregulated in the early stage (peaks at 6 weeks during HFD feeding) and downregulated in the advanced stage (2 months after HFD). Consistent with autophagy, activated mitophagy is observed via Mito-Keima fluorescence analysis in vivo, and there are decreases in mitochondrial content and the mtDNA:nDNA ratio. The potential explanation for this time-dependent mitophagy in obesity and diabetes might be a vicious cycle between mitophagy and mitochondrial dysfunction because the increase in dysfunctional mitochondria activates mitophagy for clearance in the early stage. However, defective mitophagy is responsible for the accumulation of damaged mitochondria in a later stage. However, age-related factors cannot be ruled out in defective mitophagy in the advanced phase. Interestingly, increased expression of the mitochondrial autophagy markers BCL2 interacting protein 3 (BNIP3) and p62 was revealed in response to short-term HFD feeding, but no difference in LC3II and lysosomal associated membrane protein 1 (LAMP1) levels was observed [39], indicating that there was a timely change in mitophagy initiation at the very beginning of HFD feeding. Therefore, mitophagy is classified as benign and detrimental in obesity and diabetes-related heart disorders.
- Deficient benign mitophagy in obesity and diabetes-related heart disorders
- Benign mitophagy is defined as mitophagy that is beneficial to mitochondrial maintenance, cellular homeostasis and cell viability under the pathological conditions of obesity and diabetes. This benign mitophagy is typically impaired, and there are decreased levels of autophagy and mitophagy markers [6]. In obese and diabetic rodents, excessive mtROS and mitochondrial fragmentation can be eliminated by restoring mitophagy, and mitochondrial morphology and function in cardiac tissue, as well as cardiac function, can be recovered [7,40]. Table 1 summarizes benign mitophagy in other conditions, including diabetic kidney disease, obesity-related non-alcoholic fatty liver disease (NAFLD), diabetic neuropathy [41], diabetic retinopathy, and diabetic hyposalivation.
- Excessive detrimental mitophagy in obesity and diabetes-related heart disorders
- In contrast to benign mitophagy, detrimental mitophagy is defined as mitophagy that is harmful to mitochondrial homeostasis and normal cellular activity in obesity and diabetes. Detrimental mitophagy is frequently upregulated in obesity and diabetes-related heart disorders [16], and there is an increase in the expression of autophagy and mitophagy markers [42-44]. This damaging effect is enhanced by a further increase in mitophagy but suppressed by inhibiting activated mitophagy. Furthermore, mitochondrial morphology and function are damaged due to detrimental mitophagy because this increased but dysfunctional mitophagy cannot clear damaged mitochondria. Table 2 shows detrimental mitophagy in other tissues in obesity and diabetes.
SUPPRESSED AND EXCESSIVE MITOPHAGY ARE BOTH OBSERVED IN OBESITY AND DIABETES-RELATED HEART DISORDERS
- The classical mitophagy pathway
- Similar to autophagy, mitophagy involves initiation, membrane nucleation, phagophore formation, phagophore expansion, fusion with the lysosome, and cargo degradation [11]. Mitophagy is initiated by mitochondrial fragmentation [45], and a double-layered membrane forms, followed by expansion and mitochondrial fragmentation to form a double-membraned mitophagosome. Finally, mitophagic degeneration depends on fusion with lysosomes or peroxisomes [46].
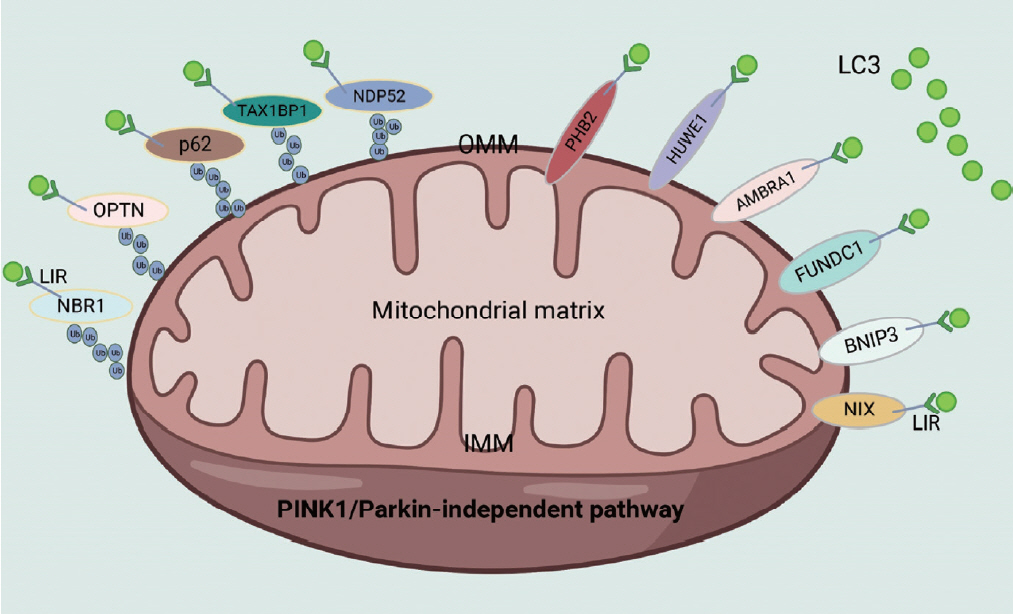
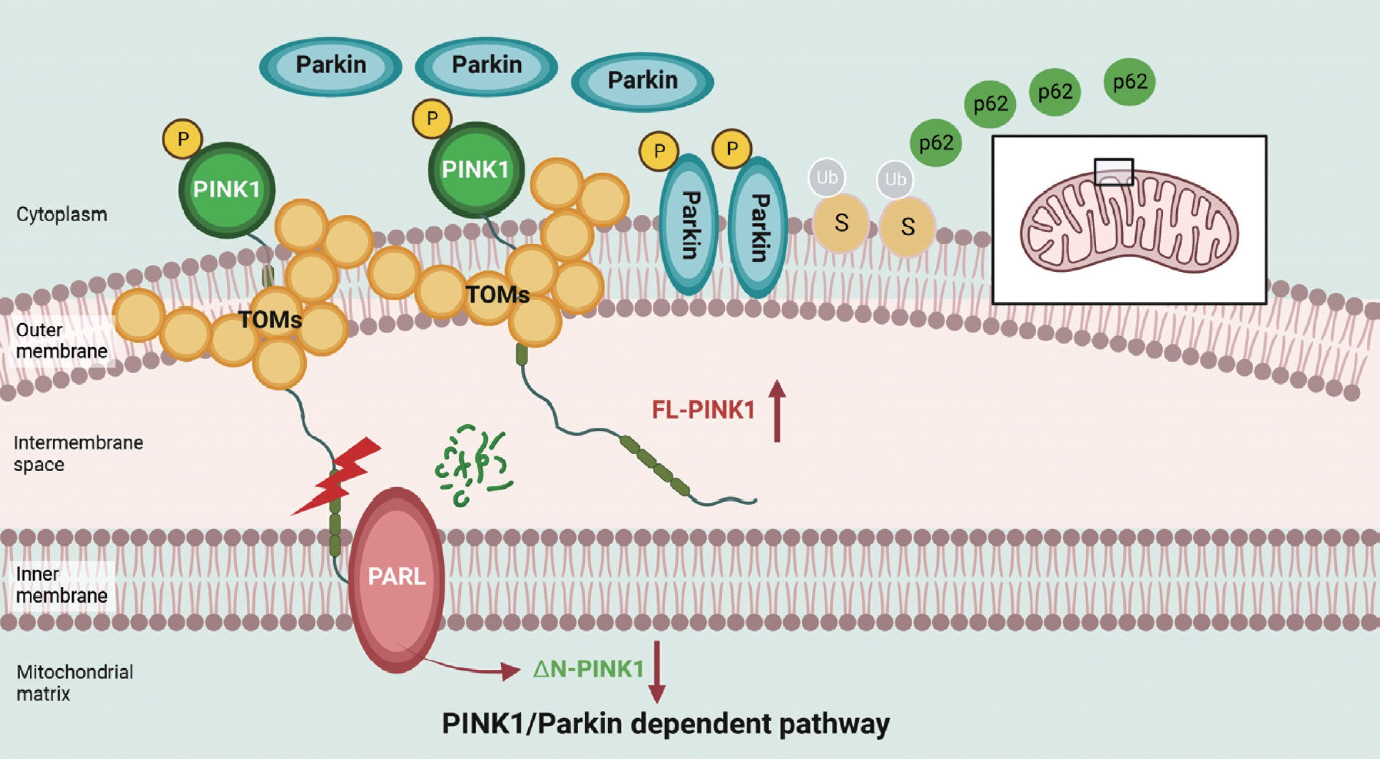
- Two classical signaling pathways exist in mammals to regulate mitophagy [1]: the PINK1/Parkin-independent and PINK1/Parkin-dependent pathways. PINK1/Parkin-independent mitophagy is characterized as receptor/adapter-mediated mitophagy (Fig. 1). Typical receptors that mediate mitophagy are almost all outer mitochondrial membrane (OMM) proteins, including FUN14 domain containing 1 (FUNDC1), BNIP3, BCL2 interacting protein 3 like (NIX), autophagy and beclin 1 regulator 1 (AMBRA1), HECT, UBA and WWE domain containing E3 ubiquitin protein ligase 1 (HUWE1), prohibitin 2 (PHB2), cardiolipin, and FKBP prolyl isomerase 8 (FKBP8). Standard adapters include p62, optineurin (OPTN), Tax1 binding protein 1 (TAX1BP1), calcium binding and coiled-coil domain 2 (NDP52), and NBR1 autophagy cargo receptor (NBR1), which can be recruited to damaged mitochondria after ubiquitination of the OMM. PINK1/Parkin-induced mitophagy mainly depends on PINK1 activation in the mitochondrial membrane and Parkin translocation from the cytoplasm to mitochondria (Fig. 2) [47]. After mitochondrial damage in response to mitochondrial stress and binding with OMM proteins such as translocase of the outer mitochondrial membrane 7 (TOM7), TOM20, TOM22, TOM40, and TOM70, the PINK1 complex is activated by kinase activity, which is essential for subsequent Parkin recruitment. An increase in Parkin translocation to mitochondria results in the ubiquitination of OMM proteins and the recruitment of P62 to these dysfunctional mitochondria to initiate mitophagy.
- Dysregulation of the mitophagy pathway in obesity and diabetes-related heart disorders
- The PINK1/Parkin-dependent mitophagy pathway and PINK1/Parkin-independent mitophagy pathway are the two classic mitophagy regulatory mechanisms and are dramatically altered in obesity and diabetes.
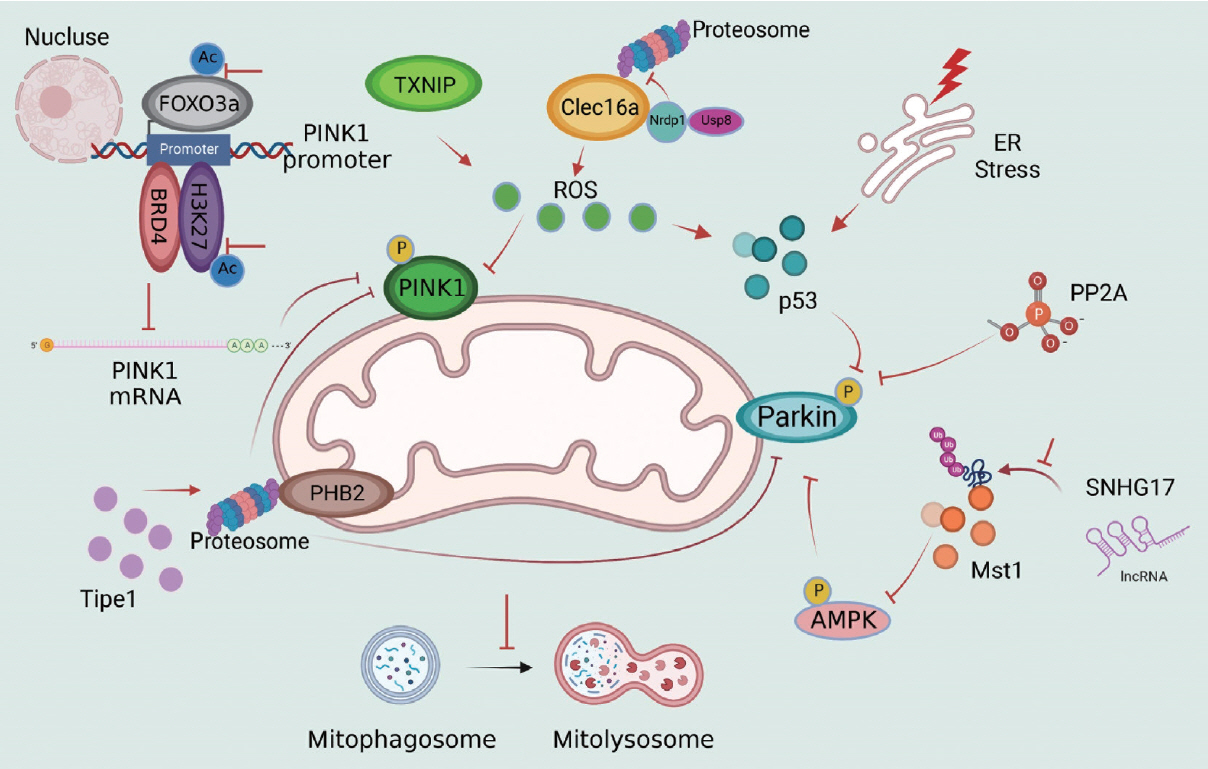
- The crucial role of PINK1 and Parkin in mitophagy regulation in obesity and diabetes has been well demonstrated. Parkin-mediated mitophagy can prevent the accumulation of dysfunctional mitochondria and consequent cardiac hypertrophy during HFD consumption [48]. In comparison, these protective effects are impaired by Parkin inhibition in Parkin-knockout (KO) diabetic mice and Parkin-small interfering RNA (siRNA) PA-treated cells [48] in vitro. PINK1 is a well-known upstream regulator of Parkin in mitophagy, and PINK1 expression is often detected in the presence of Parkin. Under physiological conditions, healthy mitochondria are polarized. Immature PINK1, which is also known as full-length PINK1 (FL-PINK1), is shuttled into the mitochondrial matrix and cleaved to form the mature form: N-terminal–cleaved PINK1 (ΔN-PINK1). As mitochondrial dysfunction occurs, PINK1 maturation becomes defective, leading to FL-PINK1 accumulation at the OMM, which is responsible for mitophagy initiation [8,49]. Moreover, this process is regulateed by forkhead box O3a (FOXO3a) acetylation and the bromodomain containing 4 (BRD4)-acetylated histone H3 lysine 27 (H3K27ac) complex, which can bind to the PINK1 promoter and regulate PINK1 messenger RNA (mRNA) expression under HF stress [50,51]. After PINK1 deletion, PINK1 and Parkin expression is reduced, and oxidative stress, inflammation, apoptosis and cell dysfunction are increased [52,53]. The impairment of PINK1/Parkin-dependent mitophagy and the potential upstream mechanisms in obesity and T2DM are illustrated in Fig. 3.
- Excessive reactive oxygen species (ROS) generation inhibits PINK1 activation, thus suppressing p-Parkin levels but not total Parkin levels in HG conditions [17,54]. Inactivation of PINK1-p-Parkin results in impaired mitophagy and defects in mtROS clearance, which forms a vicious cycle between ROS and defective mitophagy [15]. Thioredoxin-interacting protein (TXNIP) is increased in the diabetic brain, and PINK1/Parkin-mediated mitophagy is impaired [55]. After TXNIP-siRNA transfection in PC12 cells, cellular ROS production and cytochrome c oxidase subunit 4 (COX-IV)-LAMP1 colocalization were suppressed, suggesting that TXNIP regulates PINK1-mediated mitophagy and mitophagy degradation [56]. Inversely, TXNIP might be the upstream regulator of ROS, since it is involved in ROS-PINK1/Parkin-mediated mitophagy in diabetic mice via nuclear factor, erythroid derived 2, like 2 (Nrf2)/kelch-like ECH-associated protein 1 (Keap1) or Nrf2/antioxidant response element (ARE) signaling [54,57].
- C-type lectin domain family 16 member A (Clec16a), an initiator of redox and inflammatory cascades [17,58], inhibits Parkin-mediated mitophagy activation in physiological conditions by forming the Clec16a-ring finger protein 41 (Nrdp1)-ubiquitin specific peptidase 8 (Usp8) complex. As a specific partner of Clec16a, Nrdp1 directly contacts Clec16a and is protected from proteasomal degradation [59]. After mitochondrial damage during pathological states, Clec16a-Nrdp1 promotes autophagosome-lysosome fusion or mitophagosome-lysosome fusion via advanced mitophagy through Parkin activation. Furthermore, this protective effect is abolished by Clec16a pharmacologic inhibitor treatment and in Clec16a-KO mice, and there is a further increase in Parkin-mediated mitophagy and detrimental mitophagy in diabetic mice. Moreover, enhanced cytosolic p53 expression is observed due to excessive ER stress and oxidative stress and reduced Parkin-dependent mitophagy in diabetic β-cells, indicating a negative regulatory effect on mitophagy in a redox-dependent manner [60].
- Mammalian sterile 20-like kinase 1 (Mst1) has been reported to negatively regulate mitophagy via a Parkin-dependent pathway, and Mst1 can reduce Parkin expression, thereby suppressing mitophagy in diabetic cardiomyocytes [18]. After the inhibition of Mst1 phosphorylation and activity, Parkin-mediated mitophagy was enhanced in diabetic hearts [61,62]. Recently, we discovered that long non-coding RNA (lncRNA) small nucleolar RNA host gene 17 (SNHG17) was the upstream regulator of Mst1 in Parkin-mediated mitophagy. LncRNA SNHG17 overexpression led to the elevated Mst1 protein levels by inhibiting the ubiquitination and degradation of Mst1 [45] and a consequent suppression of Parkin-dependent mitophagy. In addition, Parkin-mediated mitophagy is restored after Mst1 deletion [18], indicating the crucial role of the lncRNA SNHG17-Mst1-Parkin pathway in mitophagy regulation in diabetic kidneys.
- Elevated expression of tumor necrosis factor, alpha-induced protein 8-like 1 (TIPE1) has been observed in both diabetic patients and diabetic mice. Furthermore, renal tubular epithelial cell (RTEC)-specific Tipe1 deletion attenuates cellular damage, reducing collagen accumulation, increasing epithelial cell markers, and alleviating renal fibrosis in diabetic nephropathy, which results from enhanced PINK1/Parkin-mediated mitophagy. As an inner mitochondrial membrane protein, PHB2 functions as a crucial mitophagy receptor. After Tipe1 ablation, the half-life of PHB2 was prolonged because Tipe1 could interact with PHB2 and promote the proteasomal degradation of PHB2. Moreover, the crucial role of PHB2 in Tipe1-PINK1/Parkin mitophagy has been evidenced by the fact that the cellular and mitochondrial protection of Tipe1 silencing are abolished by further PHB2 ablation [63].
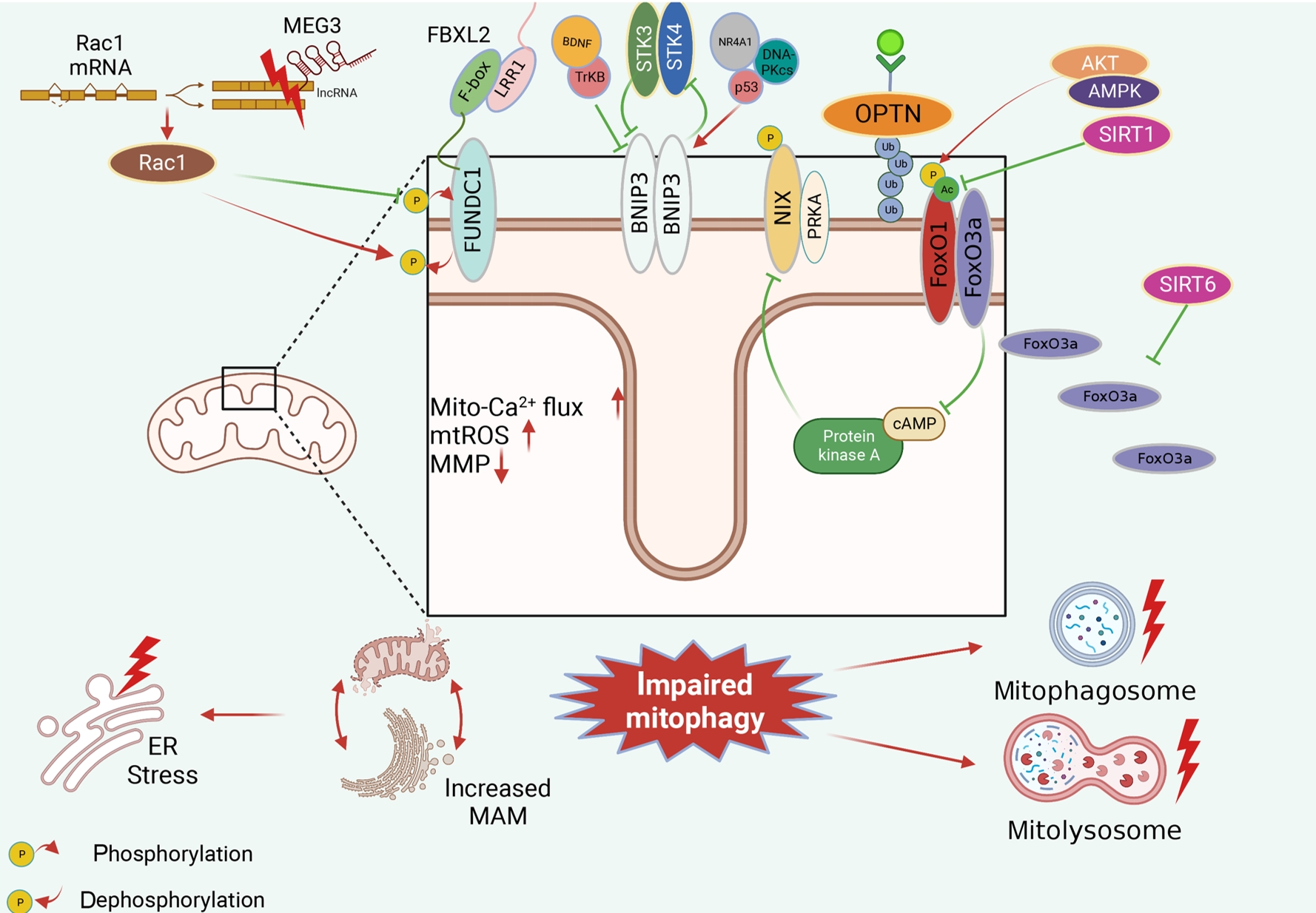
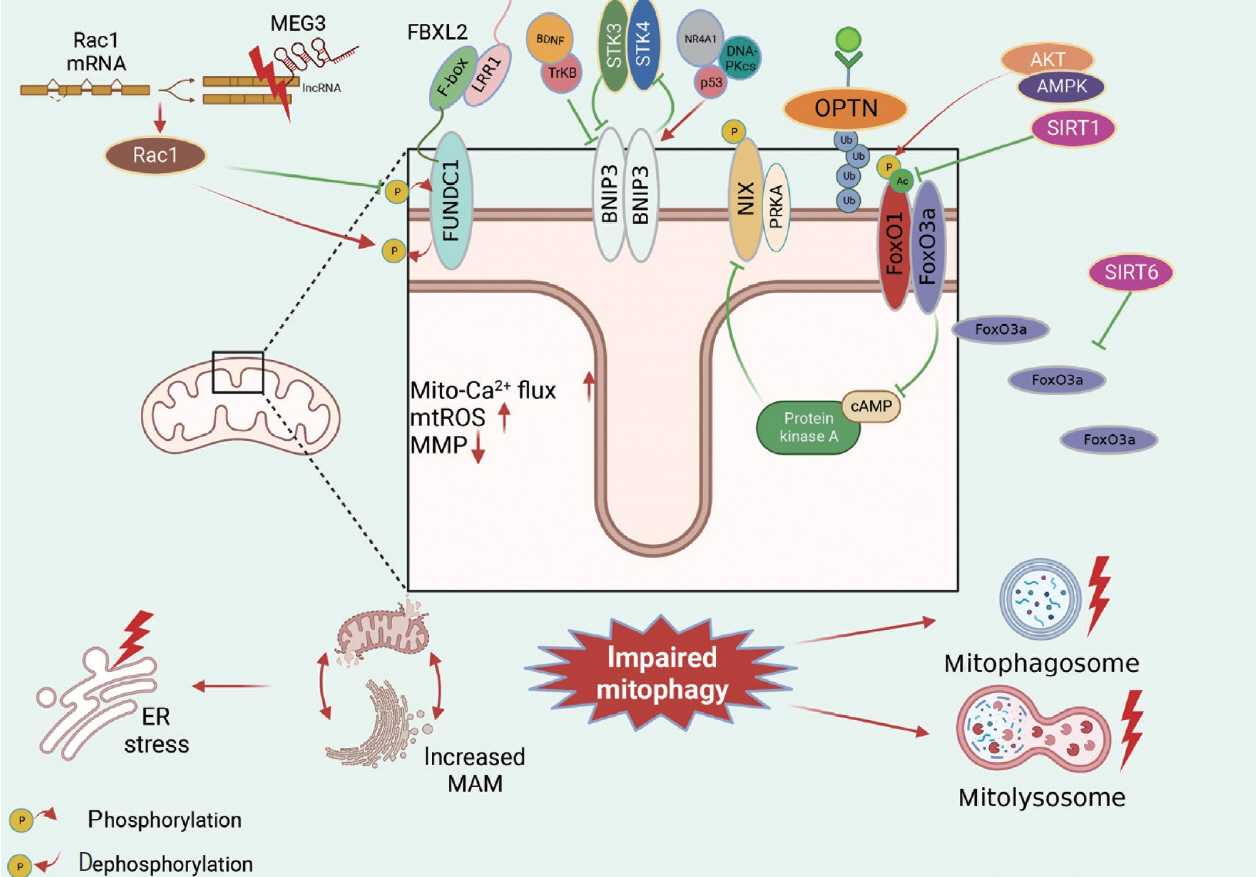
- Receptor/adapter-dependent mitophagy is another classical mitophagy. Identified mitophagy receptors/adaptors involved in obesity and diabetes include FUNDC1, BNIP3, OPTN, and FoxO1/FoxO3a (Fig. 4).
- FUNDC1 plays an essential role in mitochondrial morphology and functional regulation in obesity and diabetes. We found detrimentally elevated FUNDC1 expression in diabetic hearts. FUNDC1 overexpression induces excessive detrimental mitophagy, leading to the disruption of dysfunctional mitochondria elimination, increased mitochondrial Ca2+ flux, mitochondrial ROS overproduction, impaired mitochondrial membrane potential, and increased mitochondria-associated endoplasmic reticulum membrane (MAM) formation in diabetic hearts [19]. In contrast, FUNDC1 suppression was observed in skeletal muscle tissue during HFD feeding, which was consistent with mitophagy inhibition in diabetic mice [64], which is considered benign [31]. Maternally expressed 3 (MEG3)-Rac family small GTPase 1 (Rac1) has been reported to improve oxidative stress, inflammation and apoptosis in HG-treated neurons through FUNDC1-dependent mitophagy. MEG3 is overexpressed and can bind with the Rac1 3’ untranslated region (3’UTR); thus, inhibiting Rac1 expression and resulting in detrimental mitophagy inhibition by increasing FUNDC1 dephosphorylation in diabetic neurons, and the protective effects were abolished by FUNDC1 ablation [65]. Moreover, F-box and leucine rich repeat protein 2 (FBXL2) can directly interact with FUNDC1 and further regulate FUNDC1-mediated mitophagy, and the FUNDC1-FBXL2 interaction mainly relies on the F-box domain rather than the leucine-rich repeat protein 1 (LRR1) domain of FBXL2. However, there was no significant difference in the FUNDC1-FBXL2 interaction in cardiac tissues after low-fat and HF intake. Therefore, more research is needed to explore the potential mechanism by which the FUNDC1-FBXL2 interaction mediates FUNDC1-dependent mitophagy in obesity and diabetes [6].
- In obesity and T2DM, it is unclear whether BNIP3 expression is upregulated or downregulated; thus, whether BNIP3-mediated mitophagy is beneficial or detrimental remains ambiguous. Decreases in BNIP3 expression and BNIP3-mediated mitophagy were detected in vivo and in vitro, and mitochondrial homeostasis and cellular viability were improved after BNIP3 reactivation. BNIP3-mediated mitophagy is mediated by the brain-derived neurotrophic factor (BDNF)/tropomyosin receptor kinase B (TrkB)-dependent pathway and nuclear receptor 4A1 (NR4A1)/DNA-dependent protein kinase, catalytic subunit (DNA-PKcs)/p53 pathway, and BDNF is positively regulated by BNIP3 [66] and negatively regulated by NR4A1/DNA-PKcs/p53 activation [67]. However, we also observed that it enhanced BNIP3 expression and activated mitophagy in obesity and diabetes, which is defined as detrimental BNIP3-mediated mitophagy and occurred in a serine/threonine kinase 3 (STK3)/STK4-dependent manner. Interestingly, after BNIP3 ablation, there was a significant decrease in STK3/STK4 levels, indicating a potential feedback loop between BNIP3 and STK3/STK4 [68]. Apart from BNIP3 expression, the phosphorylation and subcellular location of BNIP3 also contribute to mitophagy regulation. Inhibitory BNIP3L phosphorylation at Ser212 within the carboxy terminus of the transmembrane domain of BNIP3L (NIX) can suppress excessive mitophagy induced by PA in myotubes. Additionally, the subcellular translocation of BNIP3 is essential for mitophagy. Increases in BNIP3 and mitochondrial marker colocalization promote mitophagy, while BNIP3 in other organelles does not affect mitophagy [69].
- Fox is a newly discovered protein family that regulates autophagy including FoxO1 and FoxO3. HFD regulates FoxO1 posttranscriptionally, including phosphorylation and acetylation, while FoxO1 expression remains unchanged. There is an increase in FoxO1 phosphorylation but no change in total FoxO1 levels in HFD-induced obesity. However, the increase in FoxO1 phosphorylation inhibits mitophagy, indicating inhibitory phosphorylation in mitophagy via the Akt kinase 2 (AKT2)/AMP-activated protein kinase (AMPK)-p-FoxO1 signaling pathway [37]. The FoxO1 acetylation level is significantly increased when mitophagy is repressed, indicating that FoxO1-mediated mitophagy is suppressed in diabetic mice. This occurs in a sirtuin 1 (Sirt1)-dependent manner, which has been proven by gain- and loss-of-function experiments [20,70].
- FoxO3 is another member of the Fox protein family that plays a vital role in mitophagy regulation in obesity and T2DM. We observed decreased levels of NIPA magnesium transporter 2 (NIPA2), a negative regulator of FoxO3a and consistently activated mitophagy in diabetic osteoblasts, which indicates that NIPA2 is involved in FoxO3a-mediated mitophagy regulation [71]. Moreover, FoxO3a cellular localization is critical for mitophagy regulation. SIRT6 activation-mediated FoxO3a translocation from the cytoplasm to mitochondria induces mitophagy, thus inhibiting mitochondrial fission in diabetic cardiomyopathy [40]. Furthermore, FoxO3 is considered an upstream regulator of other mitophagy receptors, such as BNIP3L, in obese adipocytes [72].
- In conclusion, mitophagy regulation in obesity and diabetes is complicated. All mitophagy receptors and adapters form a complex network and can regulate each other.
REGULATION OF THE MITOPHAGY PATHWAY IN OBESITY AND DIABETES-RELATED HEART DISORDERS
Impaired PINK1/Parkin-dependent mitophagy in obesity and diabetes-related heart disorders
(1) Redox pathways in PINK1/Parkin-dependent mitophagy
(2) The Mst1 pathway in PINK1/Parkin-dependent mitophagy
(3) The TIPE1-PHB2-PINK1/Parkin pathway in mitophagy
Impaired receptor/adapter-dependent mitophagy pathways in obesity and T2DM
(1) FUNDC1-mediated mitophagy
(2) BNIP3-mediated mitophagy
(3) Fox-mediated mitophagy
- Pharmacologic suppression of mitophagy
- To date, several types of studies have investigated the effects of mitophagy suppression on obesity and T2DM. When excessive detrimental mitophagy is suppressed, we observed protective effects on obesity and diabetes. It has been reported that sesamol treatment results in a significant reduction in body fat and lipid accumulation after HFD consumption. We further observed that sesamol could increase mitochondrial numbers and promote mitochondrial biogenesis, which is associated with inhibiting excessive and detrimental mitophagy via the β3-adrenergic receptor/protein kinase A (β3-AR/PKA) signaling pathway in obese adipocytes [73]. KU-596, a newly discovered treatment, is reported to attenuate diabetic peripheral neuropathy in clinical patients by improving mitochondrial bioenergetics and decreasing cellular oxidative stress. Further study showed that HFD-induced excessive mitophagy was inhibited, and there were improvements in mitochondrial morphology and function, which indicates that KU-596 inhibits excessive and detrimental mitophagy in diabetes [55].
- Pharmacologic activation of mitophagy
- Moreover, many studies have demonstrated the effects of mitophagy activation on obesity and diabetes. When defective benign mitophagy is activated, we observed improvements in mitochondrial function and morphology, which reversed cellular dysfunction. In the myocardial ischemia‒reperfusion model, reduced mitophagy and enhanced mitochondrial fission were observed in diabetic mice, and there were decreases in plasma melatonin levels. Melatonin treatment can restore impaired mitophagy, thus improving mitochondrial biogenesis and inhibiting mitochondrial fission by activating SIRT6 signaling [40]. Similar effects of melatonin have been revealed in NAFLD via the NR4A1/DNA-PKcs/p53 pathway [67]. Moreover, melatonin significantly improves the early and advanced stages of mitophagy, and increased numbers of autophagosomes engulfing mitochondria and autolysosomes engulfing mitochondria were observed in HG-treated cardiomyocytes [61] in vitro. Autophagy-related 7 conditional knockout (Atg7 cKO) mice exhibit significant mitophagy dysfunction in the context of obesity and diabetes, suggesting the potential effects of autophagy on mitophagy. Therefore, a peptide derived from a region of beclin1 protein (TAT-beclin 1), which contains 18 amino acids derived from Beclin1, was administered to diabetic mice to investigate the impact of autophagy activators on mitophagy. TAT-beclin 1 improved Parkin-independent mitophagy in diabetic cardiomyopathy, attenuating HFD-induced mitochondrial dysfunction and enhancing mitochondrial turnover [38]. Coenzyme Q10 (CoQ10) has been reported to participate in ATP production and antioxidant activity. It can reverse mitophagy impairment in obese kidney and HG-treated murine glomerular endothelial cells, ameliorate mtROS generation and mitochondrial dysfunction and improve mitochondrial dynamics by restoring the Nrf2/ARE signaling pathway [57]. Additionally, mitoQ, a mitochondria-specific antioxidant, is composed of CoQ10 and tiphenylphosphine cations. Similar renal mitochondrial protective effects have been demonstrated in mitoQ-treated diabetic mice via the Nrf2-PINK1 pathway [22]. Treatment with recombinant human progranulin (rPGRN), a secreted glycoprotein, attenuates HG-induced mitochondrial dysfunction, thereby improving mitochondrial morphology and decreasing mitochondrial fission in the diabetic kidney. These protective effects on diabetic kidneys depend on the restoration of PINK1-mediated mitophagy by activating the Sirt1-peroxisome proliferative activated receptor, gamma, coactivator 1 alpha (PGC-1α)/FoxO1 pathway [20]. Urolithin A restores defective mitophagy and improves cardiac function in the obese hearts. In addition, there is a significant increase in autophagosomes and a decrease in autolysosomes after urolithin A treatment, suggesting that urolithin A activates mitophagy by enhancing the initiation but not the later stage of mitophagy in obesity [74].
PHARMACOLOGIC INTERVENTION IN MITOPHAGY IN OBESITY AND DIABETES-RELATED HEART DISORDERS
- Many studies have suggested that obesity- and diabetes-related heart disorders result from significant mitochondrial dysfunction and subsequent mitophagy damage. However, it remains unclear how mitophagy changes under these pathological conditions. Mitophagy in obesity and diabetes occurs in a tissuespecific, time-dependent, and age-related manner, and mitophagy shows different changes in different tissues and may be different in the early and advanced stages. After pharmacologic mitophagy interventions, mitochondrial morphology and function were restored, and there were additional improvements in cell viability. Therefore, mitophagy intervention might be a new therapeutic strategy for heart disorders associated with obesity and diabetes, although previous studies have been conducted just in animal models.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
This work is supported by the National Natural Science Foundation of China (82272241, 82270392, 82070421, 82072135), 1.3.5 Project for Disciplines of Excellence (ZYJC18019), Center of Excellence-International Cooperation Initiative Grant (1391 70032), China Postdoctoral Science Foundation (2023M732462), and by Projects of Sichuan Provincial Department of Science and Technology (2022NSFSC1328, 2022NSFSC1396, 2021YJ 0135).
NOTES
-
Acknowledgements
- The authors would like to acknowledge the participants and their families for participating in the study, as well as the Biorender for figure creation.


| Diseases | Mitochondrial dysfunction | Changes of mitophagy | Mitophagy evaluation | Effects of mitophagy | Mitophagy intervention | Reference |
|---|---|---|---|---|---|---|
| Diabetic cardiomyopathy | Decreased ATP synthesis, lower ΔΨm, increased mtROS, increased mtD-NA, mitochondrial calcium | Inhibition | LC3B, p62, mt-Keima | Reduced apoptosis, inflammation, mitochondrial apoptosis, and ER stress | Activation by Parkin overexpression, inhibition by FUNDC1 ablation | [6,7] |
| Diabetic kidney disease | Impaired mitochondrial dynamics, lower ΔΨm, increased mtROS, increased mtDNA | Inhibition | TEM, LC3B+VDAC/TOM20/COX-IV, p62 | Reduced ROS production, apoptosis, reduced RTEC senescence, alleviated EMT, and fibrosis | Inhibition by OPTN siRNA, inhibition by PHB2 deletion | [15,22,23,65] |
| Obesity-related NAFLD; hepatic steatosis | Decreased ATP synthesis, impaired mitochondrial dynamics, lower ΔΨm, the increased opening rate of mPTP, increased mtROS | Inhibition | LC3B (mito-LC3B), TOM20+LAMP1 | Decreased markers of liver damage, repressed hepatic lipogenesis and fibrosis, attenuated hepatocyte ROS, inflammation, and apoptosis | Inhibition by Parkin siRNA or Parkin ablation, inhibition by Bnip3 knockdown | [18,38,70] |
| Painful diabetic neuropathy | Lower ΔΨm | Inhibition | TEM, LC3B+COX-IV, COX-IV+LAMP1 | Decreased cellular ROS and apoptosis | Inhibition by lysosome deacidificant (DC661) | [41] |
| Diabetic retinopathy | Lower ΔΨm, increased mtROS | Inhibition; activation | TEM, GFP-LC3+MitoTracker, p62 | Increased proliferation, decreased inflammation and mitochondrial apoptosis | Activation by PINK1/Parkin overexpression, inhibition by PINK1 siRNA | [57] |
| Diabetic hyposalivation | Increased mitochondrial volume, decreased ATP synthesis, lower ΔΨm, decreased mtDNA | Activation | TEM, LC3B, p62, mitochondria and lysosome markers | Improved morphology and secretion of SMG | None | [17] |
ATP, adenosine triphosphate; mtROS, mitochondrial reactive oxygen species; mtDNA, mitochondrial DNA; LC3B, microtubule-associated protein 1 light chain 3 beta; ER, endoplasmic reticulum; FUNDC1, FUN14 domain containing 1; TEM, transmission electron microscope; VDAC, voltage-dependent anion channel; TOM20, translocase of the outer mitochondrial membrane 20; COX-IV, cytochrome c oxidase subunit 4; ROS, reactive oxygen species; RTEC, renal tubular epithelial cell; EMT, epithelial-mesenchymal transition; OPTN, optineurin; siRNA, small interfering RNA; PHB2, prohibitin 2; NAFLD, non-alcoholic fatty liver disease; mPTP, mitochondrial permeability transition pore; LAMP1, lysosomal associated membrane protein 1; Bnip3, BCL2 interacting protein 3; DC661, the lysosome deacidificant; GFP-LC3, green fluorescent protein-light chain 3; PINK1, PTEN-induced putative kinase 1; SMG, submandibular gland.
| Diseases | Mitochondrial dysfunction | Changes of mitophagy | Mitophagy evaluation | Effects of mitophagy | Mitophagy intervention | Reference |
|---|---|---|---|---|---|---|
| Obesity-exposed oocytes | Decreased ATP synthesis, lower ΔΨm | Activation | PINK1 | Failure in the conversion from fertilized oocytes to the blastocyst stage in oocytes | None | [42] |
| Obesity-induced cardiomyopathy | Decreased ATP synthesis, reduced mitochondrial mass, decreased mtDNA | Activation | TEM, LC3B+TOM20, TOM20+LAMP1, mt-Keima | Increased cardiac dysfunction | Inhibition by PINK1 siRNA | [16] |
| Diabetic osteoporosis | Reduced intracellular Mg2+ | Activation | TEM, LC3B, p62, PINK1, Parkin | Impaired osteogenic capability | Inhibition by Parkin-RNAi | [74] |
| Diabetic nephropathy | Decreased ATP synthesis, impaired mitochondrial dynamics, increased mtROS | Activation | LC3B, p62, BNIP3 | Not mentioned | None | [43] |
| Diabetes-related depression | Lower ΔΨm, increased mtROS | Activation | GFP-LC3, mRFP-LC3, beclin 1, Parkin | Enhanced apoptosis, exacerbated depressionlike behavior | Activation by rapamycin, inhibition by MHY1485 | [44] |
| Diabetic retinopathy | Mitochondrial morphology alterations | Activation then inhibition | Mitophagy-reporter mice, FL-Pink1/ΔN-Pink1 ratio, LC3B+COX-IV | Increased cellular senescence | None | [8] |
ATP, adenosine triphosphate; PINK1, PTEN-induced putative kinase 1; mtDNA, mitochondrial DNA; TEM, transmission electron microscope; LC3B, microtubule-associated protein 1 light chain 3 beta; TOM20, translocase of the outer mitochondrial membrane 20; LAMP1, lysosomal associated membrane protein 1; siRNA, small interfering RNA; mtROS, mitochondrial reactive oxygen species; BNIP3, BCL2 interacting protein 3; GFP-LC3, green fluorescent protein-light chain 3; mRFP-LC3, monomeric red fluorescent protein-light chain 3; MHY1485, the mTOR receptor agonist; FL-Pink1, full-length PINK1; ΔN-Pink1, N-terminal–cleaved PINK1; COX-IV, cytochrome c oxidase subunit 4.
- 1. Li W, He P, Huang Y, Li YF, Lu J, Li M, et al. Selective autophagy of intracellular organelles: recent research advances. Theranostics 2021;11:222-56.ArticlePubMedPMC
- 2. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ 2013;20:21-30.ArticlePubMedPMCPDF
- 3. Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res 2017;120:1812-24.ArticlePubMed
- 4. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res 2020;126:1501-25.ArticlePubMedPMC
- 5. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 2016;12:144-53.ArticlePubMedPMCPDF
- 6. Ren J, Sun M, Zhou H, Ajoolabady A, Zhou Y, Tao J, et al. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv 2020;6:eabc8561.ArticlePubMedPMC
- 7. Shao D, Kolwicz SC Jr, Wang P, Roe ND, Villet O, Nishi K, et al. Increasing fatty acid oxidation prevents high-fat diet-induced cardiomyopathy through regulating parkin-mediated mitophagy. Circulation 2020;142:983-97.ArticlePubMedPMC
- 8. Hombrebueno JR, Cairns L, Dutton LR, Lyons TJ, Brazil DP, Moynagh P, et al. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight 2019;4:e129760.ArticlePubMedPMC
- 9. Koch RE, Josefson CC, Hill GE. Mitochondrial function, ornamentation, and immunocompetence. Biol Rev Camb Philos Soc 2017;92:1459-74.ArticlePubMedPDF
- 10. Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, et al. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab 2016;24:566-81.ArticlePubMedPMC
- 11. Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF. Mitophagy and Neuroprotection. Trends Mol Med 2020;26:8-20.ArticlePubMed
- 12. Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019;15:1182-98.ArticlePubMedPMC
- 13. Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016;12:689-702.ArticlePubMedPMC
- 14. Deng R, Zhang HL, Huang JH, Cai RZ, Wang Y, Chen YH, et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy 2021;17:3011-29.ArticlePubMedPMC
- 15. Han YC, Tang SQ, Liu YT, Li AM, Zhan M, Yang M, et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis 2021;12:925.ArticlePubMedPMCPDF
- 16. Wang SH, Zhu XL, Wang F, Chen SX, Chen ZT, Qiu Q, et al. LncRNA H19 governs mitophagy and restores mitochondrial respiration in the heart through Pink1/Parkin signaling during obesity. Cell Death Dis 2021;12:557.ArticlePubMedPMCPDF
- 17. Xiang RL, Huang Y, Zhang Y, Cong X, Zhang ZJ, Wu LL, et al. Type 2 diabetes-induced hyposalivation of the submandibular gland through PINK1/Parkin-mediated mitophagy. J Cell Physiol 2020;235:232-44.ArticlePubMedPMCPDF
- 18. Zhou T, Chang L, Luo Y, Zhou Y, Zhang J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkinrelated mitophagy. Redox Biol 2019;21:101120.ArticlePubMedPMC
- 19. Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, et al. Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondriaassociated endoplasmic reticulum membranes in vivo. Circulation 2019;139:1913-36.ArticlePubMedPMC
- 20. Zhou D, Zhou M, Wang Z, Fu Y, Jia M, Wang X, et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis 2019;10:524.ArticlePubMedPMCPDF
- 21. He F, Huang Y, Song Z, Zhou HJ, Zhang H, Perry RJ, et al. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J Exp Med 2021;218:e20201416.ArticlePubMedPMCPDF
- 22. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/ PINK1. Redox Biol 2017;11:297-311.ArticlePubMedPMC
- 23. Chen K, Dai H, Yuan J, Chen J, Lin L, Zhang W, et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis 2018;9:105.ArticlePubMedPMCPDF
- 24. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 2018;20:1013-22.ArticlePubMedPDF
- 25. McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, et al. Mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 2016;214:333-45.ArticlePubMedPMCPDF
- 26. Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, et al. Measuring in vivo mitophagy. Mol Cell 2015;60:685-96.ArticlePubMedPMC
- 27. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012;14:177-85.ArticlePubMedPDF
- 28. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008;454:232-5.ArticlePubMedPMCPDF
- 29. Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011;334:1144-7.ArticlePubMed
- 30. Jin X, Wang K, Wang L, Liu W, Zhang C, Qiu Y, et al. RAB7 activity is required for the regulation of mitophagy in oocyte meiosis and oocyte quality control during ovarian aging. Autophagy 2022;18:643-60.ArticlePubMedPMC
- 31. Wu H, Wang Y, Li W, Chen H, Du L, Liu D, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019;15:1882-98.ArticlePubMedPMC
- 32. Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017;552:187-93.ArticlePubMedPMCPDF
- 33. Patoli D, Mignotte F, Deckert V, Dusuel A, Dumont A, Rieu A, et al. Inhibition of mitophagy drives macrophage activation and antibacterial defense during sepsis. J Clin Invest 2020;130:5858-74.ArticlePubMedPMC
- 34. Li X, Huang L, Lan J, Feng X, Li P, Wu L, et al. Molecular mechanisms of mitophagy and its roles in neurodegenerative diseases. Pharmacol Res 2021;163:105240.ArticlePubMed
- 35. Marek-Iannucci S, Ozdemir AB, Moreira D, Gomez AC, Lane M, Porritt RA, et al. Autophagy-mitophagy induction attenuates cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 2021;6:e151981.ArticlePubMedPMC
- 36. de Maranon AM, Diaz-Pozo P, Canet F, Diaz-Morales N, AbadJimenez Z, Lopez-Domenech S, et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol 2022;53:102342.ArticlePubMedPMC
- 37. Wang S, Tao J, Chen H, Kandadi MR, Sun M, Xu H, et al. Ablation of Akt2 and AMPKα2 rescues high fat diet-induced obesity and hepatic steatosis through Parkin-mediated mitophagy. Acta Pharm Sin B 2021;11:3508-26.ArticlePubMedPMC
- 38. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 2019;124:1360-71.ArticlePubMedPMC
- 39. Ehrlicher SE, Stierwalt HD, Newsom SA, Robinson MM. Shortterm high-fat feeding does not alter mitochondrial lipid respiratory capacity but triggers mitophagy response in skeletal muscle of mice. Front Endocrinol (Lausanne) 2021;12:651211.ArticlePubMedPMC
- 40. Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu YL, et al. Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: role of SIRT6. J Pineal Res 2021;70:e12698.ArticlePubMedPDF
- 41. Yuan P, Song F, Zhu P, Fan K, Liao Q, Huang L, et al. Poly (ADPribose) polymerase 1-mediated defective mitophagy contributes to painful diabetic neuropathy in the db/db model. J Neurochem 2022;162:276-89.PubMed
- 42. Boudoures AL, Saben J, Drury A, Scheaffer S, Modi Z, Zhang W, et al. Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy. Dev Biol 2017;426:126-38.ArticlePubMed
- 43. Huang C, Yi H, Shi Y, Cao Q, Shi Y, Cheng D, et al. KCa3.1 mediates dysregulation of mitochondrial quality control in diabetic kidney disease. Front Cell Dev Biol 2021;9:573814.ArticlePubMedPMC
- 44. Liu J, Liu L, Han YS, Yi J, Guo C, Zhao HQ, et al. The molecular mechanism underlying mitophagy-mediated hippocampal neuron apoptosis in diabetes-related depression. J Cell Mol Med 2021;25:7342-53.ArticlePubMedPMCPDF
- 45. Guo F, Wang W, Song Y, Wu L, Wang J, Zhao Y, et al. LncRNA SNHG17 knockdown promotes Parkin-dependent mitophagy and reduces apoptosis of podocytes through Mst1. Cell Cycle 2020;19:1997-2006.ArticlePubMedPMC
- 46. Guan R, Zou W, Dai X, Yu X, Liu H, Chen Q, et al. Mitophagy, a potential therapeutic target for stroke. J Biomed Sci 2018;25:87.ArticlePubMedPMCPDF
- 47. Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 2018;28:R170-85.ArticlePubMedPMC
- 48. Thomas A, Marek-Iannucci S, Tucker KC, Andres AM, Gottlieb RA. Decrease of cardiac Parkin protein in obese mice. Front Cardiovasc Med 2020;6:191.ArticlePubMedPMC
- 49. Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 2011;20:867-79.ArticlePubMedPMC
- 50. Gupta P, Sharma G, Lahiri A, Barthwal MK. FOXO3a acetylation regulates PINK1, mitophagy, inflammasome activation in murine palmitate-conditioned and diabetic macrophages. J Leukoc Biol 2022;111:611-27.ArticlePubMedPDF
- 51. Mu J, Zhang D, Tian Y, Xie Z, Zou MH. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J Mol Cell Cardiol 2020;149:1-14.ArticlePubMedPMC
- 52. Ko MS, Yun JY, Baek IJ, Jang JE, Hwang JJ, Lee SE, et al. Mitophagy deficiency increases NLRP3 to induce brown fat dysfunction in mice. Autophagy 2021;17:1205-21.ArticlePubMedPMC
- 53. Zhou P, Xie W, Meng X, Zhai Y, Dong X, Zhang X, et al. Notoginsenoside R1 ameliorates diabetic retinopathy through PINK1-dependent activation of mitophagy. Cells 2019;8:213.ArticlePubMedPMC
- 54. Zhang Y, Xi X, Mei Y, Zhao X, Zhou L, Ma M, et al. High-glucose induces retinal pigment epithelium mitochondrial pathways of apoptosis and inhibits mitophagy by regulating ROS/ PINK1/Parkin signal pathway. Biomed Pharmacother 2019;111:1315-25.ArticlePubMed
- 55. Rodriguez YA, Kaur S, Nolte E, Zheng Z, Blagg BS, Dobrowsky RT. Novologue therapy requires heat shock protein 70 and thioredoxin-interacting protein to improve mitochondrial bioenergetics and decrease mitophagy in diabetic sensory neurons. ACS Chem Neurosci 2021;12:3049-59.ArticlePubMedPMC
- 56. Su CJ, Shen Z, Cui RX, Huang Y, Xu DL, Zhao FL, et al. Thioredoxin-interacting protein (TXNIP) regulates Parkin/PINK1- mediated mitophagy in dopaminergic neurons under highglucose conditions: implications for molecular links between Parkinson’s disease and diabetes. Neurosci Bull 2020;36:346-58.ArticlePubMedPMCPDF
- 57. Sun J, Zhu H, Wang X, Gao Q, Li Z, Huang H. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J Endocrinol 2019;240:445-65.Article
- 58. Sidarala V, Pearson GL, Parekh VS, Thompson B, Christen L, Gingerich MA, et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020;5:e141138.ArticlePubMedPMC
- 59. Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 2014;157:1577-90.ArticlePubMedPMC
- 60. Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc Natl Acad Sci U S A 2014;111:3116-21.ArticlePubMedPMC
- 61. Wang S, Zhao Z, Feng X, Cheng Z, Xiong Z, Wang T, et al. Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J Cell Mol Med 2018;22:5132-44.ArticlePubMedPMCPDF
- 62. Zhang M, Lin J, Wang S, Cheng Z, Hu J, Wang T, et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J Pineal Res 2017;63:e12418.ArticlePDF
- 63. Liu L, Bai F, Song H, Xiao R, Wang Y, Yang H, et al. Upregulation of TIPE1 in tubular epithelial cell aggravates diabetic nephropathy by disrupting PHB2 mediated mitophagy. Redox Biol 2022;50:102260.ArticlePubMedPMC
- 64. Fu T, Xu Z, Liu L, Guo Q, Wu H, Liang X, et al. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep 2018;23:1357-72.ArticlePubMed
- 65. Wang Z, Xia P, Hu J, Huang Y, Zhang F, Li L, et al. LncRNA MEG3 alleviates diabetic cognitive impairments by reducing mitochondrial-derived apoptosis through promotion of FUNDC1- related mitophagy via Rac1-ROS axis. ACS Chem Neurosci 2021;12:2280-307.ArticlePubMed
- 66. Jin H, Zhu Y, Li Y, Ding X, Ma W, Han X, et al. BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis 2019;24:511-28.ArticlePubMedPDF
- 67. Zhou H, Du W, Li Y, Shi C, Hu N, Ma S, et al. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res 2018;64:e12450.
- 68. Cho YK, Son Y, Saha A, Kim D, Choi C, Kim M, et al. STK3/STK4 signalling in adipocytes regulates mitophagy and energy expenditure. Nat Metab 2021;3:428-41.ArticlePubMedPDF
- 69. da Silva Rosa SC, Martens MD, Field JT, Nguyen L, Kereliuk SM, Hai Y, et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 2021;17:2257-72.ArticlePubMedPMC
- 70. Li W, Du M, Wang Q, Ma X, Wu L, Guo F, et al. FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/Parkin pathway. Endocrinology 2017;158:2155-67.ArticlePubMed
- 71. Zhao W, Zhang W, Ma H, Yang M. NIPA2 regulates osteoblast function by modulating mitophagy in type 2 diabetes osteoporosis. Sci Rep 2020;10:3078.ArticlePubMedPMCPDF
- 72. Altshuler-Keylin S, Shinoda K, Hasegawa Y, Ikeda K, Hong H, Kang Q, et al. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial clearance. Cell Metab 2016;24:402-19.ArticlePubMedPMC
- 73. Lin C, Chen J, Hu M, Zheng W, Song Z, Qin H. Sesamol promotes browning of white adipocytes to ameliorate obesity by inducing mitochondrial biogenesis and inhibition mitophagy via β3-AR/PKA signaling pathway. Food Nutr Res 2021;65:7577.ArticlePubMedPMCPDF
- 74. Huang JR, Zhang MH, Chen YJ, Sun YL, Gao ZM, Li ZJ, et al. Urolithin A ameliorates obesity-induced metabolic cardiomyopathy in mice via mitophagy activation. Acta Pharmacol Sin 2023;44:321-31.ArticlePubMedPMCPDF
 PubReader
PubReader ePub Link
ePub Link Cite
Cite