- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 46(2); 2022 > Article
-
ReviewPathophysiology Glial and Vascular Cell Regulation of the Blood-Brain Barrier in Diabetes
-
Xiaolong Li*
 , Yan Cai*, Zuo Zhang, Jiyin Zhou
, Yan Cai*, Zuo Zhang, Jiyin Zhou -
Diabetes & Metabolism Journal 2022;46(2):222-238.
DOI: https://doi.org/10.4093/dmj.2021.0146
Published online: March 18, 2022
1National Drug Clinical Trial Institution, Second Affiliated Hospital, Army Medical University, Chongqing, China
-
Corresponding author: Jiyin Zhou National Drug Clinical Trial Institution, Second Affiliated Hospital, Army Medical University, Chongqing 400037, China E-mail: zhoujiyin@gmail.com
- *Xiaolong Li and Yan Cai contributed equally to this study as first authors.
Copyright © 2022 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- As a structural barrier, the blood-brain barrier (BBB) is located at the interface between the brain parenchyma and blood, and modulates communication between the brain and blood microenvironment to maintain homeostasis. The BBB is composed of endothelial cells, basement membrane, pericytes, and astrocytic end feet. BBB impairment is a distinguishing and pathogenic factor in diabetic encephalopathy. Diabetes causes leakage of the BBB through downregulation of tight junction proteins, resulting in impaired functioning of endothelial cells, pericytes, astrocytes, microglia, nerve/glial antigen 2-glia, and oligodendrocytes. However, the temporal regulation, mechanisms of molecular and signaling pathways, and consequences of BBB impairment in diabetes are not well understood. Consequently, the efficacy of therapies diabetes targeting BBB leakage still lags behind the requirements. This review summarizes the recent research on the effects of diabetes on BBB composition and the potential roles of glial and vascular cells as therapeutic targets for BBB disruption in diabetic encephalopathy.
- Chronic hyperglycemia is a basic characteristic of diabetes and a major causative factor of various macrovascular and microvascular complications, including diabetic encephalopathy. Hyperglycemia-induced microvasculature alterations in the diabetic brain result in increased permeability and even dysfunction of the blood-brain barrier (BBB), namely diabetic BBB [1]. Diabetes also causes alterations in blood supply [2]. Neuroimaging data have revealed that diabetes patients often present with white matter damage and reduced brain volumes, which may be a consequence of BBB injury [3]. Furthermore, memory deficits have been observed in animal models of diabetes [4] that show BBB impairment accompanying pericyte loss and neuronal dysfunction [5]. It is generally believed that diabetic encephalopathy refers to a type of disease with abnormal structure and function of the central nervous system caused by diabetes. The common symptoms are impaired learning ability, decreased memory and dementia [6]. The obvious nascent morphological alteration in diabetic encephalopathy is the downregulation of tight junction proteins, which damages BBB integrity and causes higher molecular weight substances to leak through it [7]. This BBB deterioration leads to cognitive dysfunction and other types of dementia as a critical initial characteristic of diabetic encephalopathy [8].
- As a component of the BBB, highly specialized brain microvascular endothelial cells are linked by tight junctions and express several types of transporters. There is a functional polarity between the abluminal and luminal membrane surfaces of these endothelial cells. Pericytes support their function and envelop the abluminal side of the brain endothelium [9]. Proper functioning of the BBB keeps the brain free from hazardous materials in circulation, provides nutrients for brain tissues, and facilitates the excretion of detrimental compounds from the brain to the blood. The coordinated mutual interactions between endothelial cells and pericytes, astrocytes, and the basement membrane ensure normal functioning of the brain. BBB permeability is also rigorously controlled by tight junctions and metabolic barriers involving various transport systems and enzymes. However, due to its limited permeability, the BBB interferes with the delivery of remedial drugs into the brain [10].
- Tight junctions are the fundamental for BBB integrity with very low paracellular leakage, and are critical for controlling hydrophilic molecules [10]. They are composed of integral transmembrane proteins, such as occludins, claudins, endothelial cell-selective adhesion molecules, and junctional adhesion molecules [10]. Trans-membrane tight junction proteins are linked to the actin-based cytoskeleton via zonula occludens-1, zonula occludens-2, zonula occludens-3, cingulin, acute lymphoblastic leukemia-1 fusion partner in chromosome 6, and 7H6 (also named barmotin), which may also play an essential role in modulating tight junction tightness [11].
- BBB leakage or changes in transport systems play a critical role in the pathogenesis of several central nervous system disorders [12]. Numerous neuropathy models show disrupted BBB permeability. BBB impairment results in tight junction damage, increased transcytosis, changes in transport characteristics, and upregulated expression of leukocyte adhesion molecules. Despite representing different neurological diseases and being induced by diverse triggers, the mouse model of each disease displays similar alterations in mRNA expression in the BBB at subacute time points with the worst function [13]. Although there are several seemingly different potential causes of BBB impairment, common intracellular pathways modulate the structural and functional integrity of the BBB [10].
INTRODUCTION
- Streptozotocin-induced diabetes mice exhibit BBB disruption and its components. Diabetes causes alterations in several BBB transporters, such as P-glycoprotein and low-density lipoprotein receptor-related protein 1 [14-16]. The BBB in different brain areas is not equally susceptible to impairment in different types of diabetic animals. Through the comparison of a variety of animal models of diabetes, it is found that different types of diabetes represented by different types of animal models have large differences in the area of BBB destruction [17]. In streptozotocin-induced type 1 diabetes mellitus mice, the BBB injury areas are mainly frontal cortex, occipital cortex, parietal cortex, thalamus, and midbrain [1]. In type 2 diabetes mellitus model Bio-Breeding Zucker diabetic rat (BBZDR)/Wor rats, it is found that more than 84% of the brain regions show increased BBB permeability, while the cerebellum and midbrain do not change significantly [18]. In juvenile-onset diabetic rat models, the BBB permeability of hippocampus and striatum increases, but no change is observed in the frontal cortex and hypothalamus [19]. In a diet-induced obese model of type 2 diabetes mellitus, BBB damage mainly occurs in the hypothalamus and hippocampus [20]. Hippocampus and hypothalamus are relatively rare damaged in streptozotocin-induced type 1 diabetes mellitus [1]. Increased permeability of the BBB is first observed in the midbrain after 4 weeks of diabetes induced by streptozotocin, and then in the hippocampus, cortex, and basal ganglia. However, other areas show no damage even after 3 months of diabetes [7].
- Diabetes can alter the overall structure and function of the BBB. Immunohistochemistry has been used to show decreased expression of zonula occludens-1 in cerebral capillaries and increased leakage of perivascular immunoglobulin G from the blood, which indicates increased permeability in diabetes [21]. There is a substantial loss of BBB integrity in type 2 diabetes mellitus patients [22], rhesus monkeys [23], and mice [24]. Impaired BBB integrity has been reported in diabetic conditions both in vitro and in vivo [17]. BBB integrity is compromised to a certain degree in diabetes, which causes increased barrier leakage. Both capillaries and large vessels in the brain show alterations in morphology, such as thickened basal lamina due to collagen deposition, accumulated byproducts of lipid peroxidation, and deterioration of endothelial cells and pericytes [25].
- However, through the examination of the specimens of diabetic patients, it was found that the BBB structure of the prefrontal and temporal lobes did not change significantly. It shows that there is no significant change in the BBB structure in the cerebral cortex of diabetic patients [26]. A tracer was injected intravenously into the diabetes model Ins2AKITA mice to study whether the BBB permeability of diabetic mice was changed by quantifying and visualizing the tracer. The results proved that the BBB permeability of Ins2AKITA mice did not increase [27]. The variable results in animal studies may be due to the differences in diabetic models and might also depend on the time scale of the experiments and the limitations of the methods used to measure the permeability of the BBB.
- Diabetes enhances BBB leakage due to tight junction protein loss, but hyperglycemia is not the only damaging factor responsible for increased BBB leakage in diabetes. A large number of studies have shown that diabetes-related hypertension, increased oxidative stress, hyperlipidemia, and insulin resistance contribute to the development of BBB dysfunction [28-31]. The role of hormonal alterations in diabetes has long been of interest. Insulin receptor in endothelial cells can maintain the BBB structure by regulating the expression of tight junctions [32]. In a high glucose environment, the binding of insulin to its receptor on endothelial cells increases. Obesity is the primary phenotypic manifestation observed in the various leptin-deficient and leptin receptor-deficient rodents, but they also display some type 2 diabetes mellitus-like characteristics, such as hyperglycemia, glucose intolerance, and elevated plasma insulin [33]. Studies have found that leptin can reduce diabetic hyperglycemia, thereby alleviating complications caused by diabetic hyperglycemia, including BBB damage. The mechanism of leptin lowering blood sugar may be related to inhibiting the production of glucagon and corticosterone, increasing glucose uptake, and inhibiting hepatic glucose output [34].
- Increased levels of plasma matrix metalloproteinases are involved in the loss of BBB tight junction proteins and increased BBB leakage in diabetes [35]. Neuroinflammation also plays a critical role in the BBB breakdown that can lead to the fatal brain edema caused by diabetic ketoacidosis [36]. Impaired transport function and composition integrity of the brain endothelium results in BBB injury in diabetes, which is related to hyperglycemia, chronic inflammation, and oxidative stress. Downregulated expression of occludin and zonula occludens-1 is related to increased matrix metalloproteinases levels in diabetes in both rats and humans [37,38], and inhibition of matrix metalloproteinase-9 prevents increased leakage in streptozotocin-induced diabetic mice [39]. Cerebral occludin, but not zonula occludens-1 levels in streptozotocin-induced diabetic rats is significantly lower than that in wild-type rats. Therefore, diabetes modifies the levels of some structural proteins to change the molecular composition of tight junctions in cerebral tissue [40].
- Advanced glycation end-products induce vascular endothelial growth factor to upregulate the expression of matrix metalloproteinases, which in turn affect tight junction proteins and P-glycoprotein [39]. The redox-sensitive transcription factor NF-E2 related factor-2 (Nrf2) suppresses brain endothelial cell monocyte adhesion via upregulation of the redox-related mitochondrial transporter ABCB10 [24]. Vascular dysfunction in diabetes is associated with activation of protein kinase C and enhanced production of glycolytic intermediates, advanced glycation end-products, and reactive oxygen species because of increased glucose metabolism [41]. These continuous events may eventually lead to nuclear factor-κB activation, upregulated expression of adhesion molecules such as vascular cell adhesion molecule-1, activation of the small GTPase RhoA, and downregulated expression of tight junction proteins [42]. These changes are related to impairment of cognitive function. In diabetes, BBB injury is associated with increased oxidative stress and reactive oxygen species.
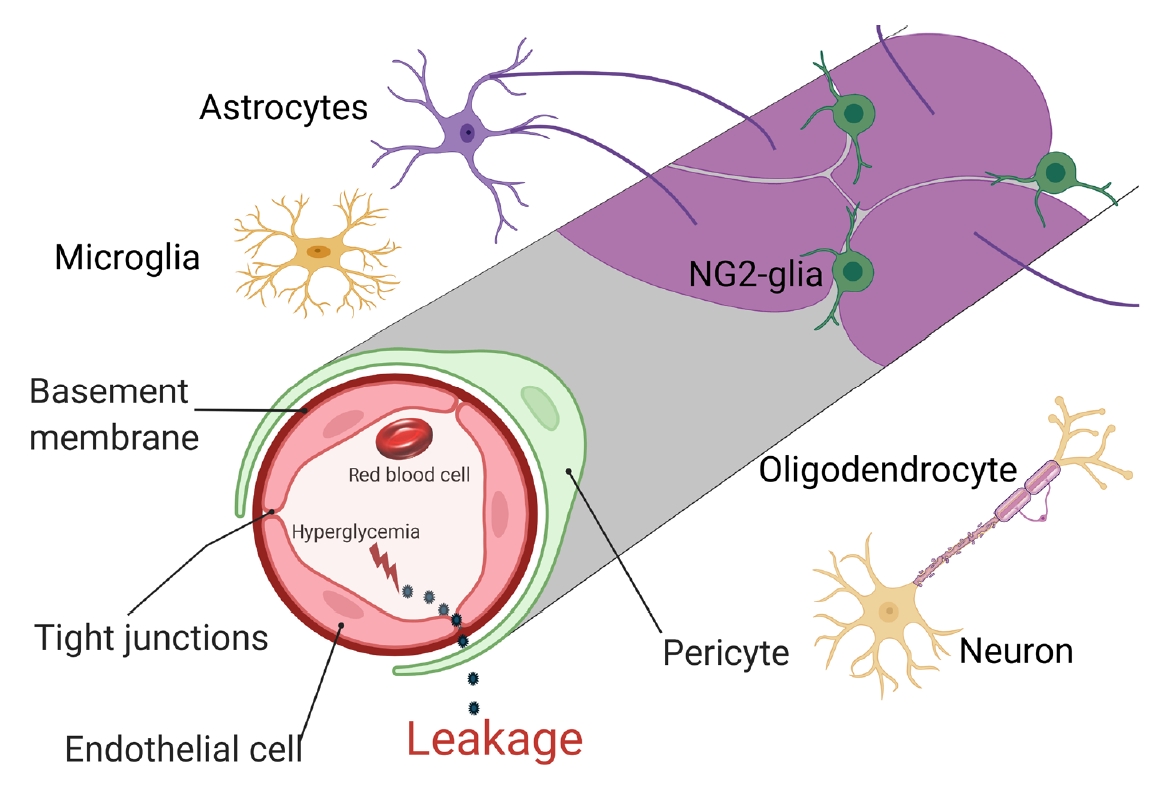
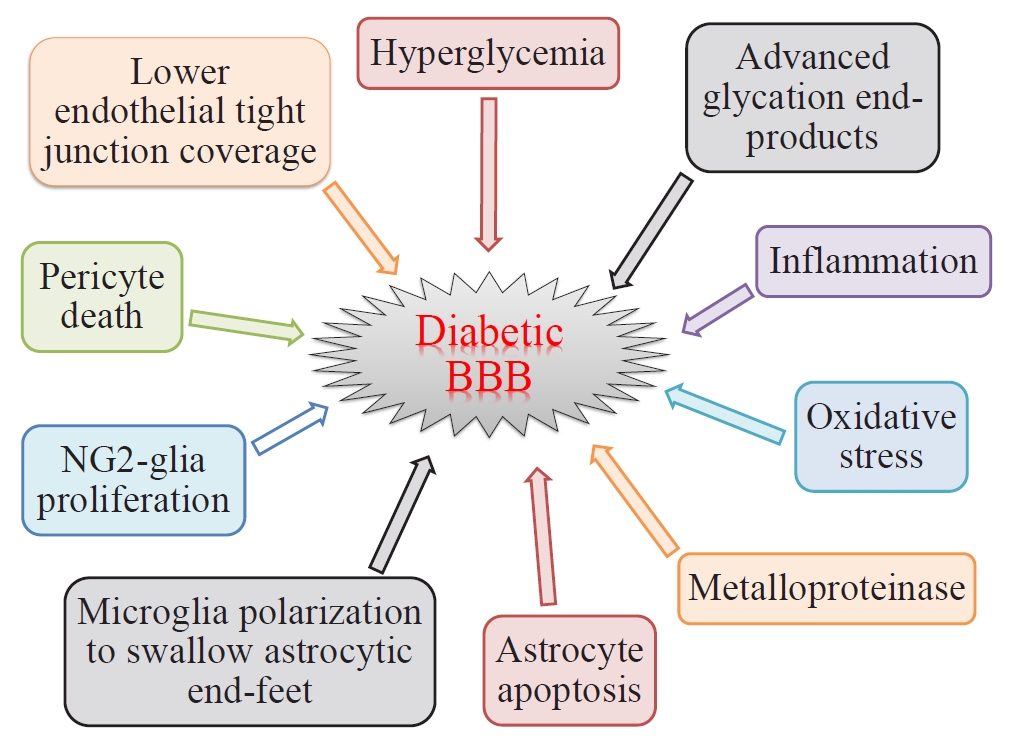
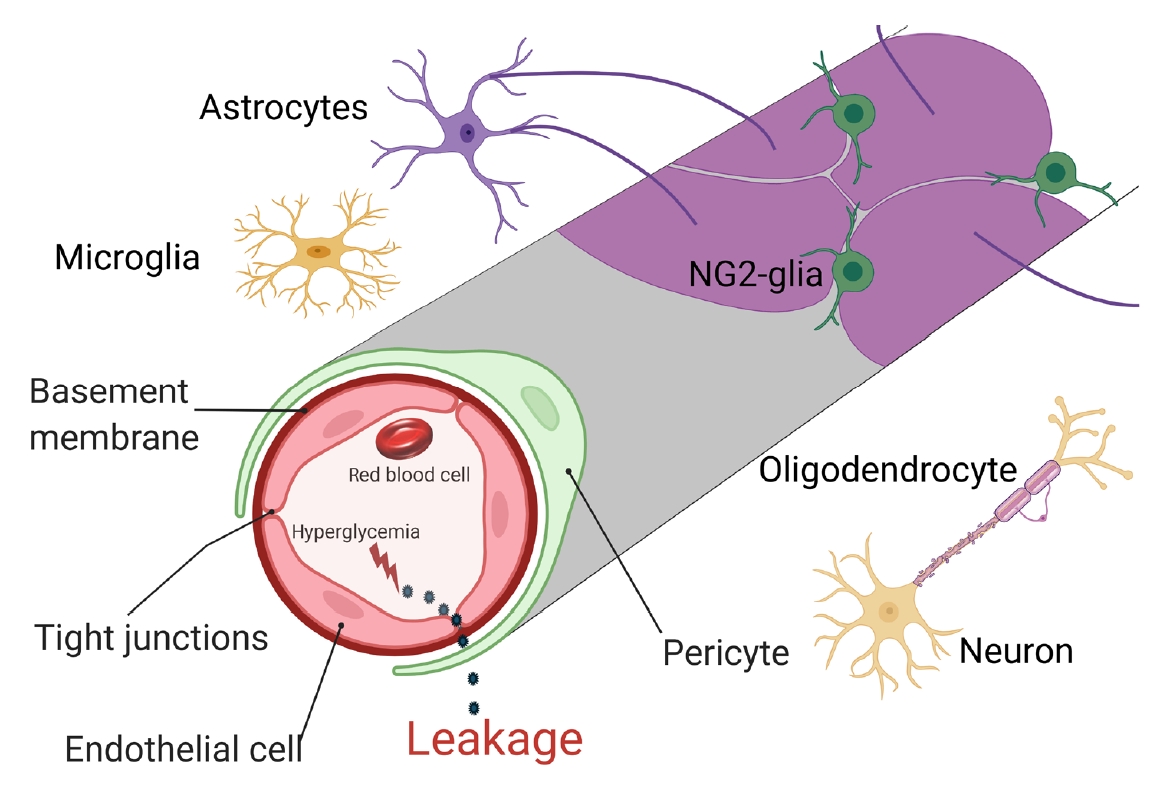
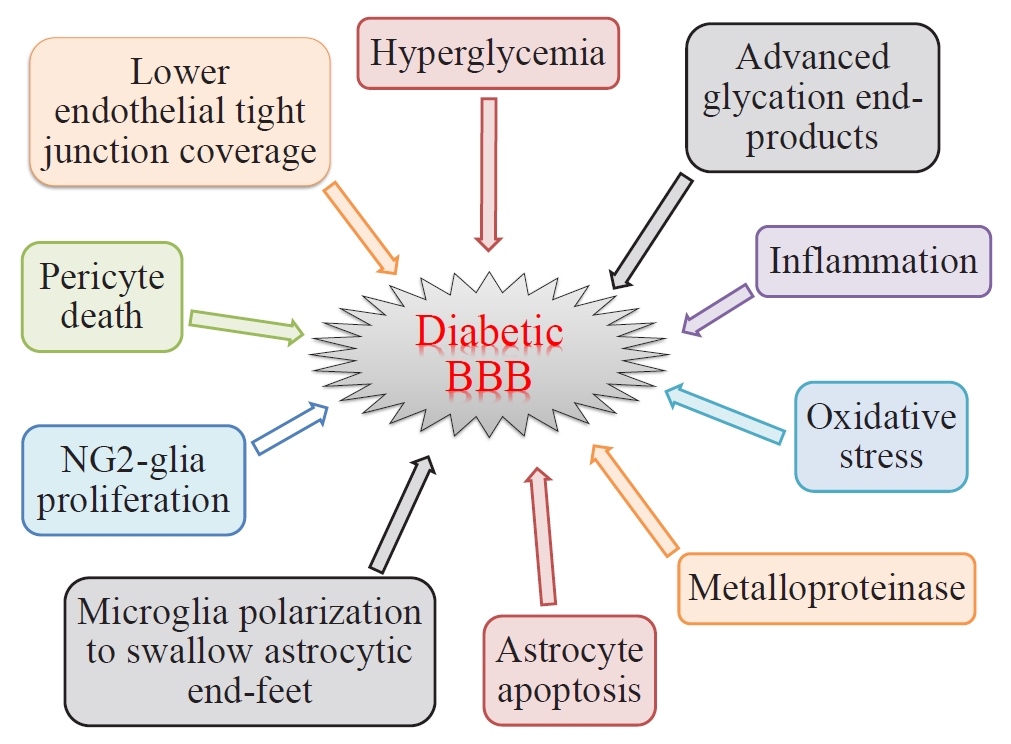
- There are two types of glia in the central nervous system: macroglia (oligodendrocytes, astrocytes, radial cells including Müller cells and Bergmann cells) and microglia. Glial neurons are equally important for the normal functioning of the nervous system. Glia are also important for protection due to their involvement in the formation of the BBB and myelin sheath development, thereby contributing to neuronal nutrition and defense mechanisms [43]. Exposing glia to hyperglycemia increases metalloproteinase 2 and metalloproteinase 9 production [44]. Fig. 1 shows the location and connection of glia (astrocytes, microglia, nerve/glial antigen 2 [NG2]-glia, and oligodendrocytes) and vascular cells (endothelial cells and pericytes) in diabetic BBB. Fig. 2 shows the detrimental effects of diabetes on glia and vascular cells that damage BBB integrity.
- Astrocytes
- Diabetes leads to astrocyte activation in the early stage of reperfusion, but causes astrocyte death in later stages. Ischemia is known to cause astrocyte activation in non-diabetic rats [45]. Moreover, increased DNA oxidation is observed before obvious astrocyte death, while augmented free radical generation is concurrent with increased astrocyte damage. These results indicate that the effects of ischemia in diabetes on astrocytes worsen with a similar efficiency as on neurons [45]. Astrocytes in the Cingulate cortex are activated by 15-minute ischemia, with stomata hypertrophy, elongated dendrites, increased number of dendrites, and upregulated expression of glial fibrillary acidic protein (GFAP), and diabetic hyperglycemia further suppresses astrocyte activation induced by ischemia [46]. Furthermore, diabetes impairs astrocytes and results in fewer astrocyte end-feet on the cerebral blood vessel wall. The activation suppression and astrocyte damage induced by diabetes may result in harmful effects on cerebral ischemia rehabilitation [46].
- As with other components of the BBB, long-term exposure of astrocytes to diabetes leads to cell-specific impairment. As shown by electron microscopy in diabetic mice, astrocytic end feet in small vessels become swollen, and the plasma membrane is detached from the basal lamina [47]. These results are linked to increased functional leakage. Serum S100B levels and/or the presence of anti-S100B autoantibodies are reliable biomarkers of BBB disruption [48]. S100B, a calcium receptor, is an astrocytic protein, and serum S100B levels in type 2 diabetes mellitus patients, but not type 1 diabetes mellitus patients, are significantly lower than those in healthy individuals. Similarly, diabetic patients and controls have very low anti-S100B autoantibody levels [49]. Chronic hyperglycemia may also impair astrocytic regulation of blood flow [50]. Astrocytes, BBB integrity, and blood flow modulation are all closely related, and the reactivity of astrocytes may be a critical element linking acute and chronic brain disorders and altered BBB permeability [51].
- In cells cultured under high-glucose conditions and brain slices of diabetic rats, communication among astrocytes via gap junctions is reduced [52]. Closely related astrocytic end-feet processes are responsible for regulating endothelial cell differentiation and inducing and maintain BBB properties. Gap junctional communication of astrocytes was found to be inhibited in brain slices and tissue cultures from diabetic rats, and reactive oxygen and nitrogen species levels are also significantly increased [52]. In vitro, the viability of human astrocytes is improved with 15 mmol/L glucose treatment, but astrocytes are damaged by 30 mmol/L glucose, with changes in the morphology and expression of cytoskeletal proteins, such as GFAP and vimentin. The levels of interleukin 6 (IL-6), IL-1, IL-4, tumor necrosis factor-α, and vascular endothelial growth factor are also increased, while that of transforming growth factor-β remained unchanged [53]. Astrocytes are essential for maintaining BBB integrity in the adult brain, and BBB-modulating factors released by other cell types, including pericytes, are not adequate to compensate for astrocyte loss [54].
- Microglia
- Microglia can also react to peripheral inflammatory disorders, which triggers them to express claudin-5 and to permeate the neurovascular unit to communicate with endothelial cells and form tight junctions to maintain BBB integrity [55]. Similarly, partial microglial deletion or inhibition of C-C motif chemokine ligand 5 signaling increases BBB leakage during the early phases of inflammation. Other factors may also cause microglia to move to blood vessels and change phenotypes under different states [55]. Lipopolysaccharide-activated microglia cause BBB dysfunction, characterized by lower trans-endothelial electrical resistance and altered expression of tight junction proteins [56].
- Chronic stress triggers microglial activation in the hippocampus through the production of IL-1β and other inflammatory cytokines and elevation of BBB leakage [57]. In co-culture of lipopolysaccharide-activated microglia with astrocytes, cytokine/chemokine dynamics are affected not only by activated microglia, but also by the crosstalk between astrocytes and activated microglia [56]. During chronic inflammation, microglia phagocytose astrocytic end-feet to damage BBB function [58]. These results suggest that microglia play a dual role in preserving BBB integrity, which has implications for clarifying how systemic immune activation affects neural functions. Activated microglia disrupt the BBB and induce the secretion of cytokines and chemokines in an in vitro model of inflammation in rats [58].
- Hypertrophic morphology and an increased number of microglia in the dorsal horn are observed in streptozotocin-induced diabetic peripheral neuropathy rats [59]. Under both high nitric oxide and hyperglycemic conditions, microglial CD11b and astrocytic GFAP expression was upregulated. Both high nitric oxide levels and hyperglycemia led to oxidative stress but not molecular changes in glia and neurons. These results indicate that high nitric oxide levels and hyperglycemia stimulated by oxidative stress may cause neuroinflammation, likely via nuclear factor-κB signaling [60]. More serious disruptions in white matter integrity are observed in diabetic patients compared with age-matched non-diabetic individuals [61]. An M2 to M1 phenotypic shift in microglia is associated with white matter injury after stroke in both young and aged mice [62]. Anti-inflammatory M2 microglia promote, whereas proinflammatory M1 microglia impair the survival of oligodendrocytes and the differentiation/maturation of NG2-glia [63].
- NG2-glia
- NG2-glia have an important role in maintaining BBB integrity [64]. Unlike other glial cells and neurons, NG2-glia express the chondroitin sulfate proteoglycan NG2. It is well known that differentiation of NG2-glia into mature oligodendrocytes is important for central nervous system development and function. In high-glucose culture media, both for 24 and 72 hours, NG2-glia have a higher differentiation rate with unaffected membrane integrity and morphology [65]. Moreover, chronic hyperglycemia causes NG2-glia to take up more glucose and release more lactate. However, unlike other glia and neurons, high-glucose exposure does not induce oxidative stress in NG2-glia [65]. These results suggest that NG2-glia can support other glia and neurons under hyperglycemic conditions.
- A momentary increase in platelet-derived growth factor receptor α-positive NG2-glia is observed in white matter injury after ischemia. However, fewer platelet-derived growth factor receptor α-positive NG2-glia are observed in db/db (diabetes) mice than in db/m mice from 4 weeks after bilateral common carotid artery stenosis [66]. These results indicate that type 2 diabetes mellitus mice exhibit more serious white matter lesions 4 weeks after chronic ischemia. Thus, a decline in the survival and proliferation of NG2-glia may play a crucial role in the development of white matter injury after ischemia in diabetes [66].
- Perivascular clustering of NG2-glia, particularly in active multiple sclerosis lesions, impairs their ability to appropriately separate from vessels following perivascular migration. NG2-glia in the perivascular space can themselves damage the BBB by disrupting astrocyte end-feet and the tight junction integrity of endothelial cells, leading to altered vascular permeability and related central nervous system inflammation [67]. Abnormal Wnt expression in NG2-glia mediates their abnormal vascular detachment and also leads to the expression of Wnt inhibitory factor-1 in NG2-glia, which disrupts the effect of Wnt ligand action on the tight junction integrity of endothelial cells [67].
- In experimental autoimmune encephalomyelitis-affected mice, NG2-glia are mainly linked to microvessels that show altered claudin-5 and occludin tight junction staining patterns and barrier permeability [64]. On the other hand, experimental autoimmune encephalomyelitis-affected NG2 knockout mice show no obvious increase in vessel-connected NG2-glia and maintain tight junctions and BBB integrity. Knockout of NG2 in NG2-glia and pericytes leads to decreased expression of vessel basal lamina molecules, such as laminin, collagen IV, and collagen VI [64].
- Oligodendrocytes
- Type 2 diabetes mellitus impairs the proliferation of NG2-glia and the generation of new myelinating oligodendrocytes [68]. Type 2 diabetes mellitus exacerbates structural injury and impairs compound action potential conduction in the white matter 35 days after stroke. The exacerbation of white matter integrity is linked to poor sensorimotor performance. Deterioration of white matter integrity and impaired oligodendrogenesis after stroke are related to poor long-range function in diabetic mice [68]. High glucose levels may switch microglia/macrophage polarization to a proinflammatory mode in microglia and NG2-glia coculture, significantly impairing NG2-glia differentiation and white matter repair. Type 2 diabetes mellitus also promotes a switch from a microglial/macrophage phenotype to a proinflammatory phenotype [68]. Glucose accelerates the differentiation of neurons and glia from embryonic neural stem cells [69], upregulates the expression of myelin mRNAs in mixed glial cultures [70], and sustains the development and myelination of oligodendrocytes in white matter slice cultures [71].
- Endothelial cells
- Diabetes-induced endothelial cell damage plays an important role, which ultimately leads to abnormal structure and function of BBB [2]. Zucker diabetic fatty rats showed significantly increased sodium fluorescein leakage and downregulated expression of tight junction proteins, such as occludin and claudin-5, in the hippocampus. Ultrastructural alterations with phagocytic discoveries in blood vessels are also observed in these mice [72]. These results indicate that long-term diabetes damages BBB permeability in the hippocampus in Zucker diabetic fatty rats by downregulating the expression of claudin-5 and occludin.
- Advanced glycation end-products damage the BBB via upregulation of matrix metalloproteinase-2 and vascular endothelial growth factor in brain microvascular endothelial cells under diabetic conditions [39]. The mRNA and protein expression of hypoxia-inducible factor 1α is upregulated in endothelial cells at high glucose levels in vitro. Moreover, as a downstream vascular effector of hypoxia inducible factor 1α, the expression of vascular endothelial growth factor is also upregulated. Vascular endothelial growth factor increases and induces the translocation of glucose transporter-1 to the cell membrane at the BBB, which promotes angiogenesis and decreases the expression of inter-endothelial tight junction proteins, such as occludin and zonula occludens-1 [73], thus causing BBB leakage. The brain microvessels of diabetic mice express lower levels of occludin than those of control mice [74]. In similar cases, downregulation of vascular endothelial growth factor expression increases the expression of occludin and zonula occludens-1 to ameliorate inter-endothelial leakage. Suppression of hypoxia-inducible factor 1α activity by its inhibitors improves BBB integrity and compactness [75].
- The potential damage to BBB integrity and permeability by hyperglycemia or hypoglycemia is mediated via altered expression/distribution of tight junction proteins and nutrient transporters in vitro. Furthermore, hypoglycemia negatively impacts the expression of oxidative and inflammatory stress markers in the endothelial cells of the BBB [76]. In the absence of changes in cell viability of the bEnd.3 endothelial cell line, bovine serum albumin and advanced glycation end-products contribute to an elevation in endothelial cell barrier permeability and a significant and prolonged oxidative stress response. Declined mitochondrial oxygen consumption is linked to these changes and may result in the generation of reactive oxygen species [77].
- BBB leakage and memory loss are linked to the decrease in tight junctions in brain endothelium and pericyte coverage, and inflammation in brain tissue in both type 1 diabetes mellitus and type 2 diabetes mellitus mice [78]. Exposure of brain microvessels to advanced glycation end-products or hyperglycemic conditions ex vivo leads to obvious abnormalities in the membranous distribution of tight junction proteins [79]. The number of extracellular vesicles isolated from diabetic mice is significantly increased, and the expression of tight junction proteins (occludin and claudin-5) is upregulated in extracellular vesicles. High glucose and advanced glycation end-product levels result in an obvious upregulation in the expression of intercellular adhesion molecules and vascular cell adhesion molecules, increased leukocyte adhesion to and migration across brain microvascular endothelial cell monolayers, and elevated BBB leakage in in vitro model with brain microvascular endothelial cells [79]. Hyperglycemia and advanced glycation end-products downregulate the expression of integrin α1, platelet-derived growth factor receptor-β1, and connexin-43 in pericytes. These data show that the BBB is damaged in ex vivo, in vitro, and in vivo models of diabetes in association with brain microvascular endothelial cells/pericyte impairment and inflammation [79].
- Pericytes
- Old diabetic rats exhibit dysregulated myogenic responses of cerebral arteries and arterioles, impaired cerebral blood flow autoregulation, increased BBB permeability, and cognitive dysfunction. These alterations are linked to the loss of vascular smooth muscle cell contractile ability, which is related to increased reactive oxygen species levels and decreased adenosine triphosphate generation [80]. Similarly, in isolated parenchymal arterioles, diminished pericyte contractile ability in diabetes is also linked to elevated reactive oxygen species levels and decreased ATP generation. Increased advanced glycation end-product production in diabetes is accompanied by lower pericyte and endothelial tight junction coverage in the cortical capillaries of old diabetic rats [81]. These results suggest that damaged cerebral hemodynamics, BBB permeability, and cognitive dysfunction in old diabetic rats are linked to hyperglycemia-triggered cerebrovascular pericyte impairment.
- Advanced glycation end-products cause hypertrophy of the basement membrane by enhancing the release of transforming growth factor-β, which has an autocrine effect to accelerate fibronectin production by pericytes. In addition, the effect of transforming growth factor-β on brain microvascular endothelial cells in conjunction with pericyte-secreted vascular endothelial growth factor and matrix metalloproteinase-2 plays a role in the altered integrity of tight junctions [39]. Advanced glycation end product-associated alterations in pericyte function can be suppressed by the siRNA for the receptor for advanced glycation end-products. Activation of the pericyte receptor for advanced glycation end-products results in further expression of the receptor [82]. Thus, advanced glycation end-products promote fibronectin production by pericytes and directly cause hypertrophy of the basement membrane in the BBB via receptors for advanced glycation end-products.
- Pericytes close to endothelial cells in cerebral microvessels are crucial to BBB integrity and are particularly vulnerable to oxidative stress, and pericytes in close apposition to the abluminal cell membrane of brain microvascular endothelial cells are found to be obviously impaired in diabetes [83]. Pericytes are basically responsible for BBB integrity, but are highly susceptible to metabolic alterations. Loss of pericytes may result in microvascular instability, causing diminished capillary perfusion in diabetic retinopathy [84]. Reduced numbers of pericytes in the diabetic mouse brain and pericyte apoptosis due to high glucose exposure are associated with oxidative stress and oxygen species production, namely, excessive superoxide production during enhanced respiration (mitochondrial oxidative metabolism of glucose) [85]. Pericyte deficiency secondary to elevated oxidative stress from redundant glucose processing via the Krebs cycle is one mechanism that results from BBB disruption [17].
- Superoxide is the precursor of all reactive oxygen species, which in turn stimulate oxidative stress. Inhibition of mitochondrial carbonic anhydrases isozymes VA and VB suppresses oxidative stress and increases pericyte numbers in the diabetic brain; it also decreases high glucose-induced respiration, oxidative stress, apoptosis, and reactive oxygen species levels in cultured brain pericytes. Cultured pericytes also show signs of oxidative stress and undergo apoptosis upon exposure to high glucose levels [85]. Diabetic mouse brains show fewer pericytes after 12 weeks of streptozotocin-induced diabetes. Advanced glycation end-products increase the production of fibronectin in pericytes via a similar upregulation of autocrine transforming growth factor-β secretion by pericytes [85]. Additionally, the suppression of mitochondrial carbonic anhydrases remarkably reduces the respiration rate and production of reactive oxygen species [37].
DIABETES DAMAGES GLIA AND VASCULAR CELLS OF BBB
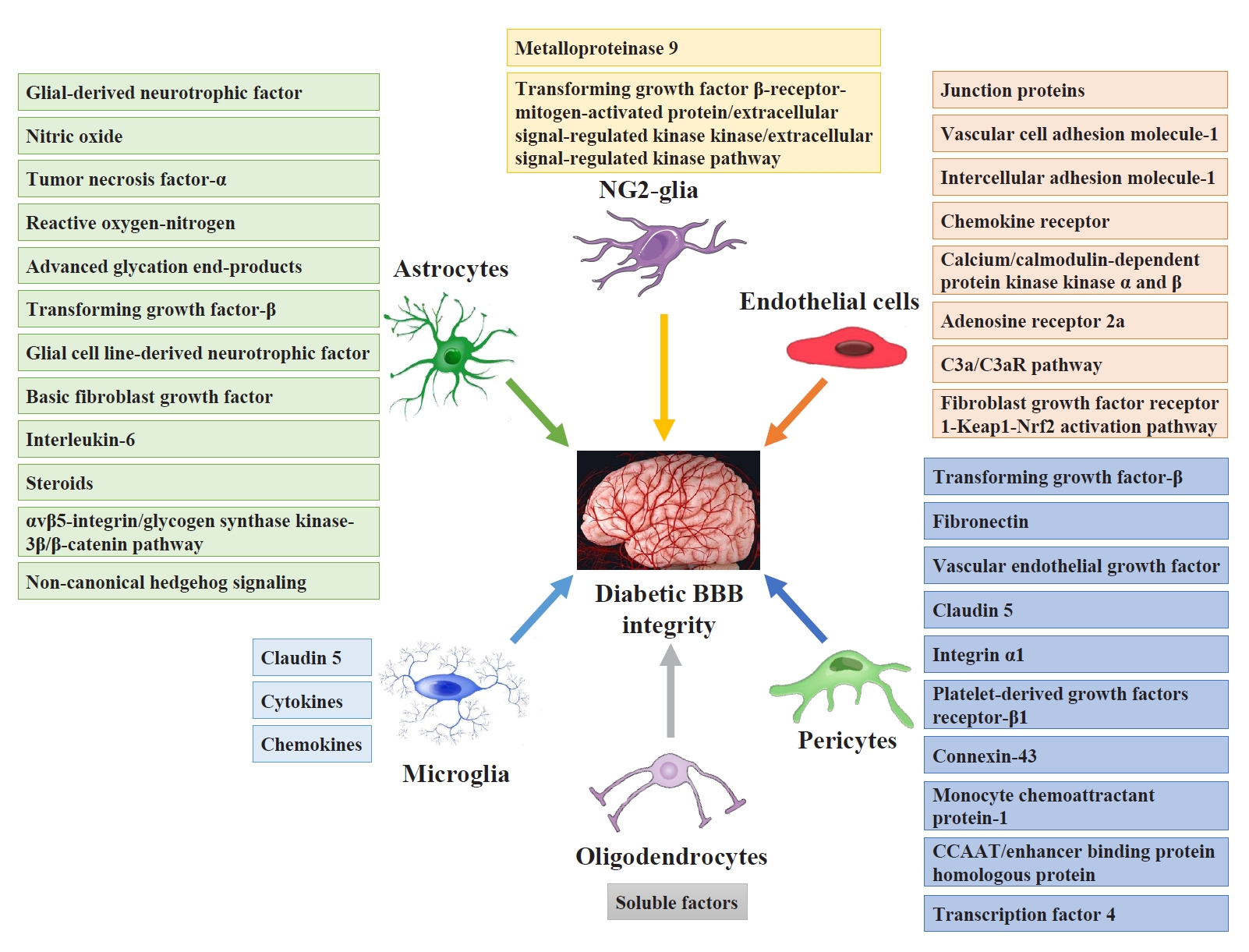
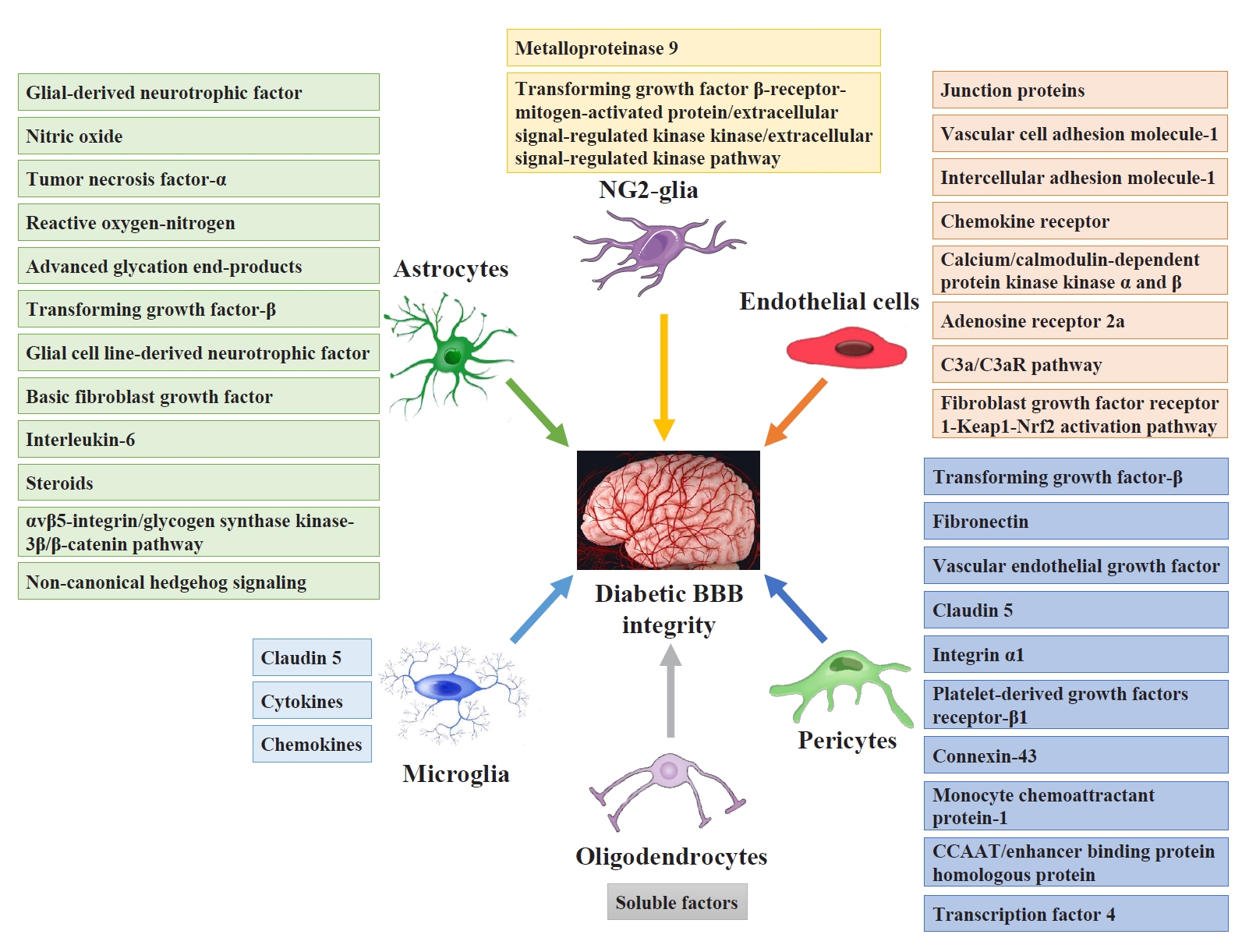
- Pericytes and astrocytes are well-known modulators of BBB maturation and integrity [86]. Pericytes are encapsulated in the basal lamina and provide direct structural support to the cerebral endothelium and metabolic support by secreting growth factors [87]. Inhibiting BBB permeability can decrease access to the brain microenvironment for a series of deleterious circulating factors, including immune cells, inflammatory cytokines, and ions. These factors activate both microglia and astrocytes directly or indirectly through neuronal lesions and aggravate neuroinflammatory impairment injury and BBB integrity [88]. Fig. 3 shows the targeting glial and vascular cells to treat diabetic BBB integrity and their mechanisms.
- Targeting astrocytes to treat diabetic BBB
- Astrocytes play a vital role in preserving normal BBB function during adult life by producing various trophic factors, including glial-derived neurotrophic factor, that maintain the BBB phenotype in endothelial cells [89]. In an in vitro co-culture model, astrocyte-derived transforming growth factor-β1 upregulated the expression of the tight junction protein zonula occludens-1 in brain microvascular endothelial cells and reinforced endothelial barrier function through non-canonical hedgehog signaling [90]. Gli2, the core transcriptional factor of the hedgehog pathway, directly regulates the expression of zonula occludens-1. The exact binding site of drosophila mothers against decapentaplegic homolog 2/3 on the gli2 promoter and of Gli2 on the zonula occludens-1 promoter were identified [90]. These results suggest that transforming growth factor-β1 could be a potential target for amelioration of BBB leakage.
- High glucose conditions cause a decline in BBB integrity and partial downregulation of zonula occludens-1, occludin, and claudin-5 in co-cultures of human brain microvascular endothelial cells and astrocytes. In human brain microvascular endothelial cell and human astrocyte co-cultures, treated with a number of antioxidants restore the damaged BBB integrity by hyperglycemia [91]. As with other cell types, high glucose-driven impairment in astrocytes is mediated by overproduction of reactive oxygen species and activation of nuclear factor-κB and signal transducer and activator of transcription 3 inflammatory pathways. Treatment with reactive oxygen species scavengers and signal transducer and activator of transcription 3 and nuclear factor-κB specific inhibitors suppresses high glucose-induced overexpression of inflammatory factors and vascular endothelial growth factor [53].
- Activated perivascular astrocytes express higher levels of GFAP, which may be a marker of BBB impairment and dysfunction [3]. Loss of astrocytes leads to vascular leakage in the retina of early diabetic mice because of increased expression of angiopoietin 2. Intravitreal injection of an angiopoietin 2 neutralizing antibody inhibits astrocyte loss and vascular leakage [92]. Angiopoietin 2 aggravates astrocyte apoptosis induced by high glucose in vitro via glycogen synthase kinase-3β activation. It binds directly to αvβ5 integrin, which is abundant in astrocytes, and the inhibition of αvβ5 integrin in vitro effectively attenuates angiopoietin 2-induced astrocyte apoptosis. Intravitreal injection of anti-αvβ5-integrin antibody inhibits astrocyte loss in early diabetic retinopathy in mice [92]. These results suggest that the angiopoietin 2-mediated astrocyte apoptosis induced by high glucose levels occurs through the αvβ5-integrin/glycogen synthase kinase-3β/β-catenin signaling pathway.
- Targeting microglia to treat diabetic BBB
- Activated microglia regulate tight junction proteins expression in endothelial cells. These tight junction proteins are vital for integrity and function of BBB [93]. Conversely, the endothelial cells also modulate the condition of microglial activation. Vessel-related microglia maintain BBB integrity through claudin-5 expression and physical communication with endothelial cells. Claudin-5 forms tight junctions with other claudin proteins and occludins near endothelial cells [94].
- Sphingosine-1-phosphate receptor 2 plays a critical role in demyelination, but not in terms of protection and myelin repair via regulating BBB permeability. Both toxin-induced and experimental autoimmune encephalomyelitis models show decreased BBB permeability and reduced numbers of Iba1+ macrophages following sphingosine-1-phosphate receptor 2 inactivation [95]. These results suggest that activation of sphingosine-1-phosphate receptor 2 inhibits remyelination and exacerbates BBB permeability and demyelination. Partial deletion of the vessel-related microglia reduces BBB permeability, as does the suppression of reactive microglia with minocycline, ameliorating BBB leakage originating from systemic inflammation [96].
- Targeting NG2-glia to treat diabetic BBB
- There is a close connection between NG2-glia and other important cells that compose of BBB. Brain endothelial cells activate Src and Akt signals within NG2-glia by releasing nutrient factors (fibroblast growth factor-2, brain-derived neurotrophic factor) to sustain NG2-glia survival and proliferation [97]. NG2-glia decrease the brain endothelial permeability and this improvement of BBB function is inhibited by a platelet-derived growth factor receptor α inhibitor, which cannot influence the oligodendrocyte-induced BBB integrity [98]. In both mouse and human brain slices, it is found that pericytes and NG2-glia are closely connected at the position around the capillaries. In vitro experiments, pericytes and NG2-glia support each other through material exchange [99]. This further shows that NG2-glia play an important role in maintaining the normal structure and function of BBB.
- NG2-glia and astrocytes are adjacent in position [100,101]. Astrocytes can secrete the protective factors of NG2-glia to protect them from oxidative stress, starvation, and oxygen-glucose deprivation [102]. Endogenous erythropoietin from astrocyte protects the NG2-glia against hypoxic and reoxygenation injury [103]. In a parental isolation mouse model, NG2-glia mediate the development of astrocytes in the hippocampus through the Wnt/β-catenin signaling pathway. The loss of NG2-glia inhibits the development and function of astrocytes [104]. In contrast to other glia and neurons, high-glucose exposure does not cause oxidative stress in NG2-glia cultures [65]. Therefore, NG2-glia may aid neurons and other glia during hyperglycemia.
- Conditioned medium from NG2-glia increases tight junction protein expression and decreases BBB leakage by activating the transforming growth factor β-receptor-mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase signaling pathway in vitro. Immuno-electron microscopy was used to demonstrate that NG2-glia connect to cerebral endothelial cells through the basal lamina in neonatal mouse brains [86]. These results demonstrate that NG2-glia improve BBB permeability by upregulating the expression of tight junction proteins through transforming growth factor-β signaling, and also that NG2-glia play a critical role in preserving BBB integrity.
- Targeting oligodendrocytes to treat diabetic BBB
- NG2-glia lower the permeability of brain endothelial cells to sodium fluorescein, but this improved BBB integrity is negated by the administration of AG1296, an inhibitor of platelet-derived growth factor receptor α [98]. Oligodendrocytes also enhance BBB integrity, but treatment with antagonists of platelet-derived growth factor receptor α does not impair oligodendrocyte-induced BBB integrity [98]. These results imply that oligodendrocytes augment BBB integrity via pathways other than those involving platelet-derived growth factor receptor-BB/platelet-derived growth factor receptor α signaling exerted by brain endothelial cell-secreted platelet-derived growth factor-BB [98]. This suggests that oligodendrocytes essentially sustain BBB integrity via soluble factors. Minocycline, a broad-spectrum antibacterial tetracycline antibiotic, facilitates white matter repair by ameliorating BBB damage, upregulating angiogenesis and the expression of tight junction proteins, and finally promoting NG2-glia proliferation to maintain oligodendrocytes in rats [105].
- Targeting endothelial cells to treat diabetic BBB
- The brain microvascular endothelial cell monolayer is linked by junctional complexes, which are composed of gap, adherent, and tight junctions. Expression and activity of histone deacetylase 3 are significantly increased in the hippocampus and cortex of db/db mice. Specific histone deacetylase 3 suppression remarkably ameliorated BBB leakage and downregulated the expression of junction proteins in db/db mice [106]. In cultured human brain microvascular endothelial cells, hyperglycemia and IL-1β treatment significantly increased transendothelial leakage and reduced the expression of junction proteins. Histone deacetylase 3 suppression remarkably attenuated transendothelial leakage and downregulated the expression of junction proteins. The potential mechanism is at least partly due to the histone deacetylase 3 suppression-mediated miR-200a/Kelch-like ECH-associating protein 1 (Keap1)/Nrf2 signaling pathway and downstream targeting junction protein expression in db/db mice [106].
- Treatment with recombinant fibroblast growth factor 21 significantly ameliorates BBB leakage and preserves the expression of junction proteins in db/db mice [107]. Recombinant fibroblast growth factor 21 also improves transendothelial permeability and junction protein downregulation induced by hyperglycemia and IL-1β in cultured human brain microvascular endothelial cells. Recombinant fibroblast growth factor 21 can activate fibroblast growth factor receptor 1, which increases its interaction with Keap1, a repressor of Nrf2, thus decreasing Keap1-Nrf2 interaction, resulting in Nrf2 secretion [107]. These results indicate that recombinant fibroblast growth factor 21 administration may decrease type 2 diabetes mellitus-induced BBB breakdown, at least partly through the fibroblast growth factor receptor 1-Keap1-Nrf2 activation pathway.
- Insulin plays an important role in diabetic encephalopathy by changing the structure and function of the BBB. In streptozotocin-induced diabetic mice, insulin increases expressions of tight junction proteins in cerebral microvessel, including occludin, claudin-5, and zonula occludens-1 [108]. Knockout adenosine receptor 2a in endothelial cells inhibits BBB leakage on high-fat diet induced obesity [109]. In human brain microvascular endothelial cells, downregulating hypoxia-inducible factor-1 activity and inhibiting its downstream gene vascular endothelial growth factor ameliorates the increased paracellular permeability and the alterations of distribution pattern of occludin and zonula occludens-1 induced by high glucose level [75]. Treatment with topiramate, a mitochondrial carbonic anhydrase inhibitor, attenuates diabetes-induced BBB breakdown and ultrastructural alterations in BBB composition [1].
- Targeting pericytes to treat diabetic BBB
- Both astrocytes [110] and pericytes [111] play important roles in BBB integrity and function. Like astrocytes, pericytes release various factors that upregulate the expression of tight junction proteins in vitro [112]. During development, pericytes aid in the development of the BBB by suppressing the expression of endothelial genes that increase vascular permeability [113] and inducing the polarization of astrocyte end-feet adjoining the vasculature [111].
- Besides their autocrine/paracrine and structural roles, brain pericyte also engage in crosstalk with brain microvascular endothelial cells, promoted in part via gap junctions between the cells. The connexin-43 protein levels in both retinas and retinal vascular cells of the diabetic rats are significantly lower than those of control ones. The number of apoptotic cells, pericyte loss, acellular capillaries, retinal vascular permeability, and thickness are obviously higher in the retinas of the diabetic and connexin-43 siRNA-treated rats than those of control ones. Downregulated connexin-43 expression leads to vascular cell death and increases vascular permeability in the retina [114]. These results indicate that diabetes-induced downregulated connexin-43 expression involves in promoting retinal vascular impairment linked to diabetic retinopathy.
- Atorvastatin promotes the maturation of BBB characteristics in new vessels by boosting endothelial tight junction formation. Proliferating NG2-positive perivascular pericytes are involved in the effects of atorvastatin on BBB maturation via modulation of endothelial tight junction strand formation in vivo and in vitro [115]. These data indicate the curative potential of atorvastatin in promoting complete BBB integrity and functional stroke recovery, and a vital role for pericyte-mediated endothelial tight junction formation in remodeling vasculature [115].
TARGETING GLIA AND VASCULAR CELLS OF BBB TO TREAT DIABETIC BBB
- BBB breakdown is a characteristic of diabetes pathology and plays an important role in diabetes-related neurological diseases. The BBB is a highly specific structure that safeguards the distinct microenvironment of the brain. Endothelial cells, linked by junctional complexes and containing many transporters, function as the main cell type in the BBB. Apart from endothelial cells, other components, such as pericytes, astrocytes, microglia, NG2-glia, oligodendrocytes, basement membrane, and perivascular macrophages, form and contribute to the proper functioning and integrity of the BBB. Diabetes causes the BBB leakage via downregulation of tight junction proteins, leading to dysfunctions of endothelial cells, pericytes, astrocytes, microglia, NG2-glia, and oligodendrocytes. More researches are still required to answer the temporal modulation, mechanisms of molecular and signaling pathways, and consequences of the BBB leakage in diabetes. So restoring functions of glial or vascular cells as therapeutic targets may ameliorate the diabetic BBB leakage. Effective therapy targeting BBB breakdown in diabetes are still awaited.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
The National Natural Science Foundation of China (No. 8177-0806), the Natural Science Foundation of Chongqing (cstc2021jcyj-msxmX0249), and Special Project for Enhancing Science and Technology Innovation Ability of Army Medical University (No. 2019XYY16) supported this work.
NOTES
-
Acknowledgements
- None
- 1. Salameh TS, Shah GN, Price TO, Hayden MR, Banks WA. Blood-brain barrier disruption and neurovascular unit dysfunction in diabetic mice: protection with the mitochondrial carbonic anhydrase inhibitor topiramate. J Pharmacol Exp Ther 2016;359:452-9.ArticlePubMedPMC
- 2. Bogush M, Heldt NA, Persidsky Y. Blood brain barrier injury in diabetes: unrecognized effects on brain and cognition. J Neuroimmune Pharmacol 2017;12:593-601.ArticlePubMedPMCPDF
- 3. Mogi M, Horiuchi M. Neurovascular coupling in cognitive impairment associated with diabetes mellitus. Circ J 2011;75:1042-8.ArticlePubMed
- 4. Sharma B, Singh N. Pitavastatin and 4’-hydroxy-3’-methoxyacetophenone (HMAP) reduce cognitive dysfunction in vascular dementia during experimental diabetes. Curr Neurovasc Res 2010;7:180-91.ArticlePubMed
- 5. Stranahan AM, Hao S, Dey A, Yu X, Baban B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J Cereb Blood Flow Metab 2016;36:2108-21.ArticlePubMedPMCPDF
- 6. Mijnhout GS, Scheltens P, Diamant M, Biessels GJ, Wessels AM, Simsek S, et al. Diabetic encephalopathy: a concept in need of a definition. Diabetologia 2006;49:1447-8.ArticlePubMedPDF
- 7. Huber JD, VanGilder RL, Houser KA. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am J Physiol Heart Circ Physiol 2006;291:H2660-8.ArticlePubMed
- 8. Ninomiya T. Diabetes mellitus and dementia. Curr Diab Rep 2014;14:487.ArticlePubMedPDF
- 9. Hill J, Rom S, Ramirez SH, Persidsky Y. Emerging roles of pericytes in the regulation of the neurovascular unit in health and disease. J Neuroimmune Pharmacol 2014;9:591-605.ArticlePubMedPMCPDF
- 10. Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Bloodbrain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 2006;1:223-36.ArticlePubMedPDF
- 11. Chen CH, Mayo JN, Gourdie RG, Johnstone SR, Isakson BE, Bearden SE. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am J Physiol Cell Physiol 2015;309:C600-7.ArticlePubMedPMC
- 12. Daneman R. The blood-brain barrier in health and disease. Ann Neurol 2012;72:648-72.ArticlePubMed
- 13. Munji RN, Soung AL, Weiner GA, Sohet F, Semple BD, Trivedi A, et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat Neurosci 2019;22:1892-902.ArticlePubMedPMCPDF
- 14. Wu KC, Pan HJ, Yin HS, Chen MR, Lu SC, Lin CJ. Change in P-glycoprotein and caveolin protein expression in brain striatum capillaries in New Zealand obese mice with type 2 diabetes. Life Sci 2009;85:775-81.ArticlePubMed
- 15. Liu H, Zhang D, Xu X, Liu X, Wang G, Xie L, et al. Attenuated function and expression of P-glycoprotein at blood-brain barrier and increased brain distribution of phenobarbital in streptozotocin-induced diabetic mice. Eur J Pharmacol 2007;561:226-32.ArticlePubMed
- 16. Liu H, Xu X, Yang Z, Deng Y, Liu X, Xie L. Impaired function and expression of P-glycoprotein in blood-brain barrier of streptozotocin-induced diabetic rats. Brain Res 2006;1123:245-52.ArticlePubMed
- 17. Banks WA. The blood-brain barrier interface in diabetes mellitus: dysfunctions, mechanisms and approaches to treatment. Curr Pharm Des 2020;26:1438-47.ArticlePubMed
- 18. Qiao J, Lawson CM, Rentrup KF, Kulkarni P, Ferris CF. Evaluating blood-brain barrier permeability in a rat model of type 2 diabetes. J Transl Med 2020;18:256.ArticlePubMedPMCPDF
- 19. Tsuneoka Y, Nishimura T, Oka JI. Fluorescein permeability of the blood-brain barrier is enhanced in juvenile- but not young adult-onset type 1 diabetes in rats. Biol Pharm Bull 2021;44:1088-92.ArticlePubMed
- 20. Salameh TS, Mortell WG, Logsdon AF, Butterfield DA, Banks WA. Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS 2019;16:1.ArticlePubMedPMCPDF
- 21. Acharya NK, Levin EC, Clifford PM, Han M, Tourtellotte R, Chamberlain D, et al. Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J Alzheimers Dis 2013;35:179-98.ArticlePubMed
- 22. Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry 2003;74:70-6.ArticlePubMedPMC
- 23. Xu Z, Zeng W, Sun J, Chen W, Zhang R, Yang Z, et al. The quantification of blood-brain barrier disruption using dynamic contrast-enhanced magnetic resonance imaging in aging rhesus monkeys with spontaneous type 2 diabetes mellitus. Neuroimage 2017;158:480-7.ArticlePubMed
- 24. Sajja RK, Prasad S, Tang S, Kaisar MA, Cucullo L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: role of ABCB10? Neurosci Lett 2017;653:152-8.ArticlePubMedPMC
- 25. Bouchard P, Ghitescu LD, Bendayan M. Morpho-functional studies of the blood-brain barrier in streptozotocin-induced diabetic rats. Diabetologia 2002;45:1017-25.ArticlePubMedPDF
- 26. Dai J, Vrensen GF, Schlingemann RO. Blood-brain barrier integrity is unaltered in human brain cortex with diabetes mellitus. Brain Res 2002;954:311-6.ArticlePubMed
- 27. Mae MA, Li T, Bertuzzi G, Raschperger E, Vanlandewijck M, He L, et al. Prolonged systemic hyperglycemia does not cause pericyte loss and permeability at the mouse blood-brain barrier. Sci Rep 2018;8:17462.ArticlePubMedPMCPDF
- 28. Baird TA, Parsons MW, Barber PA, Butcher KS, Desmond PM, Tress BM, et al. The influence of diabetes mellitus and hyperglycaemia on stroke incidence and outcome. J Clin Neurosci 2002;9:618-26.ArticlePubMed
- 29. Lee IK, Kim HS, Bae JH. Endothelial dysfunction: its relationship with acute hyperglycaemia and hyperlipidemia. Int J Clin Pract Suppl 2002;129:59-64.
- 30. Pricci F, Leto G, Amadio L, Iacobini C, Cordone S, Catalano S, et al. Oxidative stress in diabetes-induced endothelial dysfunction involvement of nitric oxide and protein kinase C. Free Radic Biol Med 2003;35:683-94.ArticlePubMed
- 31. Salmi M, Stolen C, Jousilahti P, Yegutkin GG, Tapanainen P, Janatuinen T, et al. Insulin-regulated increase of soluble vascular adhesion protein-1 in diabetes. Am J Pathol 2002;161:2255-62.ArticlePubMedPMC
- 32. Konishi M, Sakaguchi M, Lockhart SM, Cai W, Li ME, Homan EP, et al. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc Natl Acad Sci U S A 2017;114:E8478-87.ArticlePubMedPMC
- 33. Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev 2014;10:131-45.ArticlePubMedPMC
- 34. D’souza AM, Neumann UH, Glavas MM, Kieffer TJ. The glucoregulatory actions of leptin. Mol Metab 2017;6:1052-65.ArticlePubMedPMC
- 35. Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia 2007;50:202-11.ArticlePubMedPDF
- 36. Hoffman WH, Stamatovic SM, Andjelkovic AV. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res 2009;1254:138-48.ArticlePubMed
- 37. Shah GN, Morofuji Y, Banks WA, Price TO. High glucose-induced mitochondrial respiration and reactive oxygen species in mouse cerebral pericytes is reversed by pharmacological inhibition of mitochondrial carbonic anhydrases: implications for cerebral microvascular disease in diabetes. Biochem Biophys Res Commun 2013;440:354-8.ArticlePubMed
- 38. Zhong Y, Wang JJ, Zhang SX. Intermittent but not constant high glucose induces ER stress and inflammation in human retinal pericytes. Adv Exp Med Biol 2012;723:285-92.ArticlePubMedPMC
- 39. Shimizu F, Sano Y, Tominaga O, Maeda T, Abe MA, Kanda T. Advanced glycation end-products disrupt the blood-brain barrier by stimulating the release of transforming growth factor-β by pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro. Neurobiol Aging 2013;34:1902-12.ArticlePubMed
- 40. Chehade JM, Haas MJ, Mooradian AD. Diabetes-related changes in rat cerebral occludin and zonula occludens-1 (ZO1) expression. Neurochem Res 2002;27:249-52.PubMed
- 41. Dias IH, Griffiths HR. Oxidative stress in diabetes: circulating advanced glycation end products, lipid oxidation and vascular disease. Ann Clin Biochem 2014;51(Pt 2):125-7.ArticlePubMedPDF
- 42. Lu QY, Chen W, Lu L, Zheng Z, Xu X. Involvement of RhoA/ROCK1 signaling pathway in hyperglycemia-induced microvascular endothelial dysfunction in diabetic retinopathy. Int J Clin Exp Pathol 2014;7:7268-77.PubMedPMC
- 43. Mika J, Zychowska M, Popiolek-Barczyk K, Rojewska E, Przewlocka B. Importance of glial activation in neuropathic pain. Eur J Pharmacol 2013;716:106-19.ArticlePubMed
- 44. Mohammad G, Siddiquei MM. Role of matrix metalloproteinase-2 and -9 in the development of diabetic retinopathy. J Ocul Biol Dis Infor 2012;5:1-8.ArticlePubMedPMCPDF
- 45. Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes 2006;55:349-55.ArticlePubMedPDF
- 46. Jing L, Mai L, Zhang JZ, Wang JG, Chang Y, Dong JD, et al. Diabetes inhibits cerebral ischemia-induced astrocyte activation: an observation in the cingulate cortex. Int J Biol Sci 2013;9:980-8.ArticlePubMedPMC
- 47. Min LJ, Mogi M, Shudou M, Jing F, Tsukuda K, Ohshima K, et al. Peroxisome proliferator-activated receptor-γ activation with angiotensin II type 1 receptor blockade is pivotal for the prevention of blood-brain barrier impairment and cognitive decline in type 2 diabetic mice. Hypertension 2012;59:1079-88.ArticlePubMed
- 48. Kapural M, Krizanac-Bengez Lj, Barnett G, Perl J, Masaryk T, Apollo D, et al. Serum S-100beta as a possible marker of blood-brain barrier disruption. Brain Res 2002;940:102-4.PubMed
- 49. Hovsepyan MR, Haas MJ, Boyajyan AS, Guevorkyan AA, Mamikonyan AA, Myers SE, et al. Astrocytic and neuronal biochemical markers in the sera of subjects with diabetes mellitus. Neurosci Lett 2004;369:224-7.ArticlePubMed
- 50. Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia 2013;61:1939-58.ArticlePubMedPMCPDF
- 51. McConnell HL, Li Z, Woltjer RL, Mishra A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: correlation or causation? Neurochem Int 2019;128:70-84.ArticlePubMedPMC
- 52. Gandhi GK, Ball KK, Cruz NF, Dienel GA. Hyperglycaemia and diabetes impair gap junctional communication among astrocytes. ASN Neuro 2010;2:e00030.ArticlePubMedPMCPDF
- 53. Wang J, Li G, Wang Z, Zhang X, Yao L, Wang F, et al. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience 2012;202:58-68.ArticlePubMed
- 54. Heithoff BP, George KK, Phares AN, Zuidhoek IA, MunozBallester C, Robel S. Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. Glia 2021;69:436-72.ArticlePubMedPDF
- 55. Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 2012;3:1227.ArticlePubMedPDF
- 56. Shigemoto-Mogami Y, Hoshikawa K, Sato K. Activated microglia disrupt the blood-brain barrier and induce chemokines and cytokines in a rat in vitro model. Front Cell Neurosci 2018;12:494.ArticlePubMedPMC
- 57. Huang Z, Tan S. P2X7 receptor as a potential target for major depressive disorder. Curr Drug Targets 2021;22:1108-20.ArticlePubMedPDF
- 58. Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun 2019;10:5816.ArticlePubMedPMCPDF
- 59. Wodarski R, Clark AK, Grist J, Marchand F, Malcangio M. Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur J Pain 2009;13:807-11.ArticlePubMedPDF
- 60. Richa R, Yadawa AK, Chaturvedi CM. Hyperglycemia and high nitric oxide level induced oxidative stress in the brain and molecular alteration in the neurons and glial cells of laboratory mouse, Mus musculus. Neurochem Int 2017;104:64-79.ArticlePubMed
- 61. Ben Assayag E, Eldor R, Korczyn AD, Kliper E, Shenhar-Tsarfaty S, Tene O, et al. Type 2 diabetes mellitus and impaired renal function are associated with brain alterations and poststroke cognitive decline. Stroke 2017;48:2368-74.ArticlePubMed
- 62. Suenaga J, Hu X, Pu H, Shi Y, Hassan SH, Xu M, et al. White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp Neurol 2015;272:109-19.ArticlePubMedPMC
- 63. Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab 2013;33:1864-74.ArticlePubMedPMCPDF
- 64. Girolamo F, Errede M, Longo G, Annese T, Alias C, Ferrara G, et al. Defining the role of NG2-expressing cells in experimental models of multiple sclerosis: a biofunctional analysis of the neurovascular unit in wild type and NG2 null mice. PLoS One 2019;14:e0213508.ArticlePubMedPMC
- 65. da Rosa PM, Meira LA, Souza DO, Bobermin LD, Quincozes-Santos A, Leite MC. High-glucose medium induces cellular differentiation and changes in metabolic functionality of oligodendroglia. Mol Biol Rep 2019;46:4817-26.ArticlePubMedPDF
- 66. Yatomi Y, Tanaka R, Shimada Y, Yamashiro K, Liu M, MitomeMishima Y, et al. Type 2 diabetes reduces the proliferation and survival of oligodendrocyte progenitor cells in ishchemic white matter lesions. Neuroscience 2015;289:214-23.ArticlePubMed
- 67. Niu J, Tsai HH, Hoi KK, Huang N, Yu G, Kim K, et al. Aberrant oligodendroglial-vascular interactions disrupt the blood-brain barrier, triggering CNS inflammation. Nat Neurosci 2019;22:709-18.ArticlePubMedPMCPDF
- 68. Ma S, Wang J, Wang Y, Dai X, Xu F, Gao X, et al. Diabetes mellitus impairs white matter repair and long-term functional deficits after cerebral ischemia. Stroke 2018;49:2453-63.ArticlePubMedPMC
- 69. Fu J, Tay SS, Ling EA, Dheen ST. High glucose alters the expression of genes involved in proliferation and cell-fate specification of embryonic neural stem cells. Diabetologia 2006;49:1027-38.ArticlePubMedPDF
- 70. Royland JE, Konat GW, Wiggins RC. Myelin gene activation: a glucose sensitive critical period in development. J Neurosci Res 1993;36:399-404.ArticlePubMed
- 71. Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci 2011;31:538-48.ArticlePubMedPMC
- 72. Yoo DY, Yim HS, Jung HY, Nam SM, Kim JW, Choi JH, et al. Chronic type 2 diabetes reduces the integrity of the bloodbrain barrier by reducing tight junction proteins in the hippocampus. J Vet Med Sci 2016;78:957-62.ArticlePubMedPMC
- 73. Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes bloodbrain barrier breakdown. Proc Natl Acad Sci U S A 2009;106:1977-82.ArticlePubMedPMC
- 74. Vorbrodt AW, Dobrogowska DH, Tarnawski M, Meeker HC, Carp RI. Immunogold study of altered expression of some interendothelial junctional molecules in the brain blood microvessels of diabetic scrapie-infected mice. J Mol Histol 2006;37:27-35.ArticlePubMedPDF
- 75. Yan J, Zhang Z, Shi H. HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell Mol Life Sci 2012;69:115-28.ArticlePubMedPDF
- 76. Sajja RK, Prasad S, Cucullo L. Impact of altered glycaemia on blood-brain barrier endothelium: an in vitro study using the hCMEC/D3 cell line. Fluids Barriers CNS 2014;11:8.ArticlePubMedPMCPDF
- 77. Dobi A, Rosanaly S, Devin A, Baret P, Meilhac O, Harry GJ, et al. Advanced glycation end-products disrupt brain microvascular endothelial cell barrier: the role of mitochondria and oxidative stress. Microvasc Res 2021;133:104098.ArticlePubMed
- 78. Rom S, Zuluaga-Ramirez V, Gajghate S, Seliga A, Winfield M, Heldt NA, et al. Hyperglycemia-driven neuroinflammation compromises BBB leading to memory loss in both diabetes mellitus (DM) type 1 and type 2 mouse models. Mol Neurobiol 2019;56:1883-96.ArticlePubMedPDF
- 79. Rom S, Heldt NA, Gajghate S, Seliga A, Reichenbach NL, Persidsky Y. Hyperglycemia and advanced glycation end products disrupt BBB and promote occludin and claudin-5 protein secretion on extracellular microvesicles. Sci Rep 2020;10:7274.ArticlePubMedPMCPDF
- 80. Wang S, Lv W, Zhang H, Liu Y, Li L, Jefferson JR, et al. Aging exacerbates impairments of cerebral blood flow autoregulation and cognition in diabetic rats. Geroscience 2020;42:1387-410.ArticlePubMedPMCPDF
- 81. Liu Y, Zhang H, Wang S, Guo Y, Fang X, Zheng B, et al. Reduced pericyte and tight junction coverage in old diabetic rats are associated with hyperglycemia-induced cerebrovascular pericyte dysfunction. Am J Physiol Heart Circ Physiol 2021;320:H549-62.ArticlePubMed
- 82. Wang S, Cao C, Chen Z, Bankaitis V, Tzima E, Sheibani N, et al. Pericytes regulate vascular basement membrane remodeling and govern neutrophil extravasation during inflammation. PLoS One 2012;7:e45499.ArticlePubMedPMC
- 83. Warmke N, Griffin KJ, Cubbon RM. Pericytes in diabetes-associated vascular disease. J Diabetes Complications 2016;30:1643-50.ArticlePubMed
- 84. Kim LA, Wong LL, Amarnani DS, Bigger-Allen AA, Hu Y, Marko CK, et al. Characterization of cells from patient-derived fibrovascular membranes in proliferative diabetic retinopathy. Mol Vis 2015;21:673-87.PubMedPMC
- 85. Patrick P, Price TO, Diogo AL, Sheibani N, Banks WA, Shah GN. Topiramate protects pericytes from glucotoxicity: role for mitochondrial CA VA in cerebromicrovascular disease in diabetes. J Endocrinol Diabetes 2015;2:1-7.
- 86. Seo JH, Maki T, Maeda M, Miyamoto N, Liang AC, Hayakawa K, et al. Oligodendrocyte precursor cells support blood-brain barrier integrity via TGF-β signaling. PLoS One 2014;9:e103174.ArticlePubMedPMC
- 87. Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci 2011;14:1398-405.ArticlePubMedPMCPDF
- 88. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007;10:1387-94.ArticlePubMedPDF
- 89. Igarashi Y, Utsumi H, Chiba H, Yamada-Sasamori Y, Tobioka H, Kamimura Y, et al. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem Biophys Res Commun 1999;261:108-12.ArticlePubMed
- 90. Fu J, Li L, Huo D, Zhi S, Yang R, Yang B, et al. Astrocyte-derived TGFβ1 facilitates blood-brain barrier function via noncanonical hedgehog signaling in brain microvascular endothelial cells. Brain Sci 2021;11:77.ArticlePubMedPMC
- 91. Allen CL, Bayraktutan U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and bloodbrain barrier hyperpermeability. Diabetes Obes Metab 2009;11:480-90.ArticlePubMed
- 92. Yun JH, Park SW, Kim JH, Park YJ, Cho CH, Kim JH. Angiopoietin 2 induces astrocyte apoptosis via αvβ5-integrin signaling in diabetic retinopathy. Cell Death Dis 2016;7:e2101.ArticlePubMedPMCPDF
- 93. da Fonseca AC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, et al. The impact of microglial activation on bloodbrain barrier in brain diseases. Front Cell Neurosci 2014;8:362.ArticlePubMedPMC
- 94. Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the bloodbrain barrier in a focal cerebral ischemic insult. J Mol Neurosci 2011;44:130-9.ArticlePubMedPDF
- 95. Seyedsadr MS, Weinmann O, Amorim A, Ineichen BV, Egger M, Mirnajafi-Zadeh J, et al. Inactivation of sphingosine-1-phosphate receptor 2 (S1PR2) decreases demyelination and enhances remyelination in animal models of multiple sclerosis. Neurobiol Dis 2019;124:189-201.ArticlePubMed
- 96. Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation 2015;12:26.ArticlePubMedPMCPDF
- 97. Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci 2009;29:4351-5.ArticlePubMedPMC
- 98. Kimura I, Dohgu S, Takata F, Matsumoto J, Watanabe T, Iwao T, et al. Oligodendrocytes upregulate blood-brain barrier function through mechanisms other than the PDGF-BB/ PDGFRα pathway in the barrier-tightening effect of oligodendrocyte progenitor cells. Neurosci Lett 2020;715:134594.ArticlePubMed
- 99. Maki T, Maeda M, Uemura M, Lo EK, Terasaki Y, Liang AC, et al. Potential interactions between pericytes and oligodendrocyte precursor cells in perivascular regions of cerebral white matter. Neurosci Lett 2015;597:164-9.ArticlePubMedPMC
- 100. Butt AM, Ibrahim M, Ruge FM, Berry M. Biochemical subtypes of oligodendrocyte in the anterior medullary velum of the rat as revealed by the monoclonal antibody Rip. Glia 1995;14:185-97.ArticlePubMed
- 101. Orthmann-Murphy JL, Abrams CK, Scherer SS. Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci 2008;35:101-16.ArticlePubMedPMCPDF
- 102. Arai K, Lo EH. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res 2010;88:758-63.ArticlePubMedPMC
- 103. Kato S, Aoyama M, Kakita H, Hida H, Kato I, Ito T, et al. Endogenous erythropoietin from astrocyte protects the oligodendrocyte precursor cell against hypoxic and reoxygenation injury. J Neurosci Res 2011;89:1566-74.ArticlePubMed
- 104. Wang Y, Su Y, Yu G, Wang X, Chen X, Yu B, et al. Reduced oligodendrocyte precursor cell impairs astrocytic development in early life stress. Adv Sci (Weinh) 2021;8:e2101181.ArticlePubMedPDF
- 105. Yang Y, Kimura-Ohba S, Thompson JF, Salayandia VM, Cosse M, Raz L, et al. Vascular tight junction disruption and angiogenesis in spontaneously hypertensive rat with neuroinflammatory white matter injury. Neurobiol Dis 2018;114:95-110.ArticlePubMedPMC
- 106. Zhao Q, Zhang F, Yu Z, Guo S, Liu N, Jiang Y, et al. HDAC3 inhibition prevents blood-brain barrier permeability through Nrf2 activation in type 2 diabetes male mice. J Neuroinflammation 2019;16:103.ArticlePubMedPMCPDF
- 107. Yu Z, Lin L, Jiang Y, Chin I, Wang X, Li X, et al. Recombinant FGF21 protects against blood-brain barrier leakage through Nrf2 upregulation in type 2 diabetes mice. Mol Neurobiol 2019;56:2314-27.ArticlePubMedPDF
- 108. Sun YN, Liu LB, Xue YX, Wang P. Effects of insulin combined with idebenone on blood-brain barrier permeability in diabetic rats. J Neurosci Res 2015;93:666-77.ArticlePubMed
- 109. Yamamoto M, Guo DH, Hernandez CM, Stranahan AM. Endothelial Adora2a activation promotes blood-brain barrier breakdown and cognitive impairment in mice with diet-induced insulin resistance. J Neurosci 2019;39:4179-92.ArticlePubMedPMC
- 110. Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol 2005;25:25-39.ArticlePubMedPDF
- 111. Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature 2010;468:557-61.ArticlePubMedPDF
- 112. Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem 2004;89:503-13.ArticlePubMed
- 113. Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010;468:562-6.ArticlePubMedPMCPDF
- 114. Tien T, Muto T, Barrette K, Challyandra L, Roy S. Downregulation of connexin 43 promotes vascular cell loss and excess permeability associated with the development of vascular lesions in the diabetic retina. Mol Vis 2014;20:732-41.PubMedPMC
- 115. Yang Y, Yang LY, Salayandia VM, Thompson JF, Torbey M, Yang Y. Treatment with atorvastatin during vascular remodeling promotes pericyte-mediated blood-brain barrier maturation following ischemic stroke. Transl Stroke Res 2021;12:905-22.ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
- Gut microbiota and type 2 diabetes mellitus: a focus on the gut-brain axis
Yi Pan, Tong Bu, Xia Deng, Jue Jia, Guoyue Yuan
Endocrine.2024; 84(1): 1. CrossRef - Long-Term Exposure of Cultured Astrocytes to High Glucose Impact on Their LPS-Induced Activation
Ayna Abdyeva, Ekaterina Kurtova, Irina Savinkova, Maksim Galkov, Liubov Gorbacheva
International Journal of Molecular Sciences.2024; 25(2): 1122. CrossRef - Role of autophagy in angiogenic potential of vascular pericytes
Soheil Zamen Milani, Aysa Rezabakhsh, Mohammad Karimipour, Leila Salimi, Narges Mardi, Maryam Taghavi Narmi, Fatemeh Sadeghsoltani, Ferzane Valioglu, Reza Rahbarghazi
Frontiers in Cell and Developmental Biology.2024;[Epub] CrossRef - The NG2-glia is a potential target to maintain the integrity of neurovascular unit after acute ischemic stroke
Xiaoyan Hu, Panpan Geng, Xiaoyun Zhao, Qian Wang, Changqing Liu, Chun Guo, Wen Dong, Xinchun Jin
Neurobiology of Disease.2023; 180: 106076. CrossRef - Tight junction disruption and the pathogenesis of the chronic complications of diabetes mellitus: A narrative review
Ma Ludivina Robles-Osorio, Ernesto Sabath
World Journal of Diabetes.2023; 14(7): 1013. CrossRef - Function and therapeutic value of astrocytes in diabetic cognitive impairment
Fanyu Meng, Jiafeng Fu, Lin Zhang, Mengqing Guo, Pengwei Zhuang, Qingsheng Yin, Yanjun Zhang
Neurochemistry International.2023; 169: 105591. CrossRef - Exploring the molecular targets for Type 2 diabetes-induced Alzheimer’s disease through bioinformatics analysis
Lin Gao, Chengyu Huang, Hui Li, Shidi Wu, Xiaoyan Zhou, Changjiang Ying
Epigenomics.2023; 15(11): 619. CrossRef - In vivo retinal imaging is associated with cognitive decline, blood-brain barrier disruption and neuroinflammation in type 2 diabetic mice
May Majimbi, Samuel McLenachan, Michael Nesbit, Fred K. Chen, Virginie Lam, John Mamo, Ryu Takechi
Frontiers in Endocrinology.2023;[Epub] CrossRef - Diabetic microvascular disease in non-classical beds: the hidden impact beyond the retina, the kidney, and the peripheral nerves
Dídac Mauricio, Mònica Gratacòs, Josep Franch-Nadal
Cardiovascular Diabetology.2023;[Epub] CrossRef - Transcranial photobiomodulation improves insulin therapy in diabetic microglial reactivity and the brain drainage system
Shaojun Liu, Dongyu Li, Tingting Yu, Jingtan Zhu, Oxana Semyachkina-Glushkovskaya, Dan Zhu
Communications Biology.2023;[Epub] CrossRef - NG2‐glia crosstalk with microglia in health and disease
Zuo Zhang, Xiaolong Li, Hongli Zhou, Jiyin Zhou
CNS Neuroscience & Therapeutics.2022; 28(11): 1663. CrossRef - Accelerated amyloid angiopathy and related vascular alterations in a mixed murine model of Alzheimer´s disease and type two diabetes
Maria Vargas-Soria, Juan Jose Ramos-Rodriguez, Angel del Marco, Carmen Hierro-Bujalance, Maria Jose Carranza-Naval, Maria Calvo-Rodriguez, Susanne J. van Veluw, Alan W. Stitt, Rafael Simó, Brian J. Bacskai, Carmen Infante-Garcia, Monica Garcia-Alloza
Fluids and Barriers of the CNS.2022;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite