- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 37(3); 2013 > Article
-
ReviewPathophysiology Neonatal Diabetes Caused by Activating Mutations in the Sulphonylurea Receptor
- Peter Proks
-
Diabetes & Metabolism Journal 2013;37(3):157-164.
DOI: https://doi.org/10.4093/dmj.2013.37.3.157
Published online: June 14, 2013
Henry Wellcome Centre for Gene Function, Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, UK.
- Corresponding author: Peter Proks. Henry Wellcome Centre for Gene Function, Department of Physiology, Anatomy and Genetics, University of Oxford, Parks Road, Oxford OX1 3PT, UK. peter.proks@dpag.ox.ac.uk
Copyright © 2013 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Adenosine triphosphate (ATP)-sensitive potassium (KATP) channels in pancreatic β-cells play a crucial role in insulin secretion and glucose homeostasis. These channels are composed of two subunits: a pore-forming subunit (Kir6.2) and a regulatory subunit (sulphonylurea receptor-1). Recent studies identified large number of gain of function mutations in the regulatory subunit of the channel which cause neonatal diabetes. Majority of mutations cause neonatal diabetes alone, however some lead to a severe form of neonatal diabetes with associated neurological complications. This review focuses on the functional effects of these mutations as well as the implications for treatment.
- Neonatal diabetes mellitus (NDM) is a form of diabetes that is diagnosed under the age of 6 months. The most common causes of neonatal diabetes are activating mutations in the adenosine triphosphate (ATP)-sensitive potassium (KATP) channel. This review discusses functional effects and therapy implications of NDM mutations in the regulatory subunit of the KATP channel-sulphonylurea receptor-1 (SUR1).
INTRODUCTION
- Neonatal diabetes mellitus (NDM) is defined as hyperglycemia that presents within the first 6 months of life. It is a rare disorder that can be either permanent throughout life (permanent NDM, PNDM) or transient (transient NDM, TNDM), with a period of remission.
- TNDM accounts approximately for 50% cases of neonatal diabetes (ND) and affects approximately one in 100,000 live births. The majority (about 80%) of cases of TNDM are caused by abnormalities of an imprinted locus on chromosome 6q24 that results in the overexpression of a paternally expressed gene [1]. Most of remaining cases are caused by mutations in either KCNJ11 or ABCC8 genes that encode the ATP-sensitive potassium (KATP) channel respectively [2]. The precise mechanism of TNDM is unknown; it has been proposed that it could be due to either a reduced insulin requirement at the time of remission or because of some compensation effects at the level of the β-cell, pancreas, or whole body [3].
- PND may be either isolated or form part of a syndrome, such as Wolcott-Rallinson syndrome due to mutations in the EIF2AK3 gene, pancreatic agenesis due to mutations in IPF-1 gene and ND with cerebellar agenesis due to mutations in the PTF-1A gene [4]. The most common cause of isolated PNDM are mutations in the genes that encode insulin (INS) and the KATP channel (KCNJ11 and ABCC8).
NEONATAL DIABETES
- KATP channels act as metabolic sensors, coupling the metabolism of a cell to its membrane potential and electrical excitability. Their activity is primarily regulated by intracellular adenosine nucleotides, with ATP having an inhibitory and magnesium adenosine diphosphate (MgADP) or magnesium adenosine triphosphate (MgATP) a stimulatory effect on channel activity. KATP channels are expressed in many tissues including pancreas, skeletal and smooth muscle, and the brain. In these tissues, KATP channels play a multitude of physiological roles [5]; however, their physiological role has been best characterized in the pancreatic β-cell.
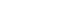
- The β-cell KATP channel links glucose metabolism and insulin release [6]. At substimulatory glucose concentrations (Fig. 1A), KATP channels are open. Hence the cell membrane is hyperpolarised, voltage-gated calcium channels are closed and no insulin is released. When blood glucose concentration rises, glucose is transported into pancreatic β-cells and metabolised, thereby increasing the ratio of ATP:adenosine diphosphate (ADP) concentrations (Fig. 1B). This closes the KATP channel, produces a membrane depolarisation that opens voltage-gated calcium channels; resulting in influx of calcium into the β-cell which triggers insulin exocytosis.
- Given the crucial role of the β-cell KATP channel in insulin secretion and glucose homeostasis, it is not surprising that mutations in this channel can lead to diseases of both hypoglycaemia and hyperglycaemia [7]. Loss of function mutations in KATP channels cause over-secretion of insulin and result in hyperinsulinaemia. Conversely, gain of function mutations cause undersecretion of insulin, hyperglycemia and result in ND.
- The β-cell KATP channel is a hetero-octameric complex comprising four Kir6.2 subunits (encoded by the KCNJ11 gene) and four sulphonylurea receptor-1 (SUR1) subunits (encoded by the ABCC8 gene). Kir6.2 is an inwardly rectifying K-channel that forms the potassium-selective pore and possesses an inhibitory site for ATP [8-10]. SUR1 is a member of the ATP binding cassette (ABC) superfamily [11]. This subunit plays multiple regulatory roles [10-13]. It confers channel sensitivity to stimulation by Mg-nucleotides, activation by K+ channel openers, such as diazoxide; and inhibition by sulphonylureas. In addition, SUR1 also enhances the inhibitory effect of ATP and stabilizes the open state of the channel in the absence of nucleotides.
THE β-CELL KATP CHANNEL
- The SUR1 protein contains three transmembrane domains (TMDs) linked by the cytosolic linker region and two nucleotide-binding domains (NBDs) (Fig. 2). TMD1 and TMD2 contain six transmembrane helices and TMD0, sited at the hydrophobic N-terminus and important for interactions with Kir6.2, contains five transmembrane helices. Each of the NBDs contains sequence motifs called Walker A and Walker B that are essential for binding the phosphate groups of nucleotides. ATP binding to SUR1 causes head to tail dimerization of the NBDs and formation of two nucleotide binding sites (NBS1 and NBS2) within the dimer interface. NBS2 possesses greater ATPase activity than NBS1 and its occupancy by MgADP stimulates KATP channel activity [14].
- Mutations in the SUR that cause ND were first identified in 2006 [15,16]. Most of patients with SUR1 mutations have isolated diabetes; approximately 30% of patients have additional neurological features. These include developmental delay, learning difficulties, epilepsy, minor dystonia, tonic posturing, and muscle weakness [17,18]. These features are consistent with the expression of KATP channels containing the SUR1 subunit in the central nervous system [9,19]. To date, only two SUR1 mutations (F132L [15,18] and I49F [20]) were identified that cause the most severe form of ND with developmental delay and epilepsy (DEND syndrome). One mutation (L213R [16]) caused ND with developmental delay but without epilepsy-intermediate DEND (i-DEND) syndrome. Unlike in some other cases of i-DEND syndrome, mutation L213R did not cause muscle weakness.
- ND mutations in SUR1 are all missense mutations and account for more than 10% of PNDM and a frequent cause of TNDM [21]. In contrast to mutations in Kir6.2 which are all dominant heterozygous, SUR1 mutations can be either dominant or recessively inherited [18]. Recessive mutations could be homozygous, mosaic due to segmental uniparental isodisomy or compound heterozygous for another activating mutation or if the second allele is inactivated. Approximately 50% of SUR1 mutations are spontaneous, arising de novo during embryogenesis [22]. To date, over 60 mutations in SUR1 have been identified; they are scattered throughout the protein sequence, but are particularly concentrated in TMD0 and their connecting loops, in the CL3 linker connecting NBD0 with NBD1 and in NBD2 (Fig. 2). The most commonly occurring mutations in SUR1 are at positions R1183 and R1380, both of which cause TNDM [22].
- Not all gain of function mutations in SUR1 result in ND, for example, mutation S1369A (the most important polymorphism of the ABCC8 gene) merely increases risk for the type 2 diabetes [23].
SUR1 AND NEONATAL DIABETES
- Gain of function mutations in the SUR1 subunit increase KATP current in the presence of MgATP. In general, there is a good correlation between the magnitude of KATP current and disease severity; thus mutations that cause largest increase KATP channel lead to the most severe form of ND with associated neurological complications-DEND syndrome [15,18]. To date, available functional studies show that increase in current produced by mutations in SUR1 is smaller than that caused by Kir6.2 mutations [14,15,18]. This may explain the relative high incidence of TNDM than PNDM among patients with ABCC8 mutations as well as why most patients with DEND syndrome (>90%) have mutations in Kir6.2.
- SUR1 mutations mediate their effect on channel activity via two main mechanisms-by reducing the inhibition produced by ATP binding at Kir6.2 and by enhancing channel activation by Mg-nucleotides.
- Mutations in SUR1 that decrease the amount of inhibition at Kir6.2 may do so in one of two ways. Firstly, they could reduce ATP binding directly. It is well established that the presence of SUR1 enhances ATP inhibition at Kir6.2, which suggests that SUR1 either contributes to the ATP binding site itself, or influences it allosterically [10]. Disruption of this could reduce ATP binding directly; mutations in SUR1 that are likely to act via this mechanism include A30V in TMD0 and G296R in TMD1 which together form a compound heterozygous mutation that results in PNDM [24]. Alternatively, SUR1 mutations can also disrupt ATP inhibition indirectly by impairing channel gating. These mutations stabilize the open state of the channel and thus increase its single channel open probability. This impairs channel ability to close both in the absence and presence of nucleotides [25]. Examples include F132L in TMD0 [15,26], L213R in the CL3 linker between TMD0 and TMD1 [27] and V324M in TMD1 [28].
- Among the mutations that enhance the activatory effect of Mg-nucleotides, many are found in NBD2 [18,29-31]. Only one SUR1-PNDM mutation (R826W) is found in NBD1 [32], nevertheless this mutation lies in the linker that is predicted to form part of NBS2. Functional studies showed that these mutations enhance the time that NBS2 spends in the activating MgADP-bound state either directly by enhancing MgADP binding [31] or indirectly by altering other reaction steps of the ATPase [29,32]. Some ND mutations which enhance Mg-nucleotide activation are located outside the NBDs [16,28,33]. These mutations may exert their effect via enhancing the transduction of the Mg-nucleotide stimulation from the NBSs of SUR1 to the channel pore at Kir6.2; alternatively, they may also allosterically affect nucleotide handling at the NBDs.
- In addition to affecting channel activity, some SUR1 mutations can also affect channel expression at the plasma membrane. An example of such mutation is V324M, of which the activating effect (enhanced Mg-nucleotide activation, enhanced stability of channel open state) is dampened by reduced channel expression at the cell surface [28]. The interplay between the two types of opposing defects may be responsible for the fact that the V324M mutation results only in the transient form of the disease.
- In general, there is no correlation between the position of the mutation in the SUR1 and the clinical phenotype-mutations in the same residue may result in different types of disease. For example, mutations of the conserved glutamate residue E1506 in NBD2 can result in either hyperinsulinism or ND [30].
- It is important to point out that several SUR1 mutations have been identified in patients with ND which were later found not to be responsible for the disease [14]. In some cases, no functional effects have been found, in others the parents were nonsymptomatic carriers of the mutation. This emphasizes the need of the functional study in order to confirm that the mutation is the cause of the disease.
FUNCTIONAL EFFECTS OF SUR1 MUTATIONS
- Prior to the discovery that ND can be caused by mutations in the KATP channel, many patients were assumed to be suffering from early-onset type 1 diabetes. Accordingly they were treated with insulin injections. Recognition that these patients actually possess gain of function mutations in KATP channel genes rapidly led to a switch to sulphonylurea treatment. Fortunately, since sulphonylureas had been used to safely treat patients with type 2 diabetes for many years, no clinical trials were required.
- Sulphonylurea drugs bind to both Kir6.2 and SUR1 subunits of the KATP channel with very different affinities [34]. The low-affinity binding site, which lies on Kir6.2, is of no clinical relevance as it is occupied only at concentrations much higher than those found in the plasma of patients treated with sulphonylureas. The primary effect of the drug is mediated via a high-affinity binding site on SUR1 with associated IC50 values for block of wild-type KATP channels being substantially lower than the usual therapeutic plasma concentrations of sulphonylureas [35].
- Depending on the type of sulphonylurea, the maximal extent of high-affinity block in the absence of nucleotides is only 50% to 80%. In the presence of nucleotides, binding of sulphonylureas to the high affinity site suppresses the activatory effect of MgADP and unmasks the inhibitory effect of nucleotides at Kir6.2. This leads to an apparent increase in the high-affinity block of wild-type KATP channels in the presence of Mg-nucleotides, which becomes virtually complete inside pancreatic β-cells [34,36].
- Thus, sulphonylureas exert dual action on KATP channel activity: a direct block and indirect block via modulation of nucleotide-dependent channel gating. Given their molecular mechanism of action, many ND mutations in SUR1 can be expected to affect sulphonylurea inhibition. Mutations that impair ATP inhibition indirectly via stabilizing the open state of the channel will reduce both direct and indirect block of the drug. Mutations that reduce ATP binding to the inhibitory site on Kir6.2 directly will impair Mg-nucleotide-induced enhancement of the high-affinity sulphonylurea block. Finally, mutations that enhance MgADP activation may potentially impair the ability of sulphonylureas to suppress it.
- Relatively little information is available at present about the efficacy of sulphonylurea block of KATP channels with ND mutations in the SUR1 subunit. Impaired direct and indirect block of sulphonylureas has been demonstrated for mutation L213R, which disrupts gating [27]. As expected, direct block was unaffected by SUR1 mutations that enhance Mg-nucleotide activation [16,33] or directly reduce ATP binding to Kir6.2 [24]. Functional studies further suggest that mutations that enhance Mg-nucleotide activation may have either no effect or cause small reduction in the indirect block of sulphonylureas [16,29,32,33,37]. The latter is likely to be mediated via displacement of sulphonylureas from SUR1 by Mg-nucleotide binding to NBS2 [38]. In support of this idea, two ND mutations that enhance ATP affinity for NBS2 were found to enhance glibenclamide displacement from SUR1 by the nucleotide [31].
- To date, most patients with ND caused by mutations in SUR1 were successfully transferred to sulphonylureas [16,33,39-43]. Major exceptions are patients with mutations in phenylalanine 132-F132L and F132V [15,18,39,40]. Mutation F132L strongly impairs channel function via altering channel gating and causes the most severe form of the disease-DEND syndrome [15,18,26]; as demonstrated by previous studies on ND mutations in Kir6.2, successful transfer to sulphonylurea therapy of patients with this syndrome is quite rare [22,44]. It is currently unclear why patients with F132V mutation that causes PNDM alone are unable to transfer to sulphonylureas. Even more puzzling is that in the only other reported case of DEND syndrome due to mutations in SUR1 (I49F), the patient was able to transfer to sulphonylurea therapy [20]. Further studies are needed to gain insight into the functional effects of these two mutations.
- An important contributing factor for successful transfer to sulphonylurea treatment is the age of the patient [39]; the sooner the transfer is carried out the better. Studies of mouse models of ND suggest that this could be due to gradual loss of β-cell mass which can be prevented by therapy [45].
- Successful treatment of patients with ND demands higher doses of sulphonylureas compared to those with type 2 diabetes [39]. As discussed above, this may be caused, at least in part, due to the fact that many ND mutations impair sulphonylurea block. Additional factors, such as reduced β-cell mass can also contribute.
- Patients with mutations in SUR1 require lower doses of sulphonylureas than those with mutations in Kir6.2 [39,44], consistent with the idea that the former mutations cause in general milder impairment of channel function than the latter. In spite of relatively high doses, the only side effects reported include transitory diarrhea [39] or tooth discoloration in a few patients [46].
- Transfer to sulphonylurea treatment does not only improve quality of life of patients with ND; it also appears to enhance their blood glucose control. Fluctuations in blood glucose are reduced and there is also a decrease in the hemoglobin A1c (HbA1c) levels [39,47]. This improvement in glycemic control is predicted to reduce the risk of diabetic complications [48,49].
SULPHONYLUREA THERAPY
- The discovery that mutations in SUR1 can cause ND has led to more insight into the role of this regulatory subunit in the KATP channel function and transformed therapy for patients with these mutations. Nevertheless, many questions remain to be answered. More functional analysis as well as high resolution crystal structure of the whole KATP channel complex are necessary in order obtain detail understanding how the SUR1 ATPase works and how it is coupled to KATP channel gating. Further studies are required to identify binding sites for therapeutic drugs. Finally, mouse models of ND will help us to understand why some mutations in SUR1 result in remitting-relapsing form of ND and why others cause additional neurological problems.
CONCLUSIONS
-
Acknowledgements
- The author would like to thank Dr. Heidi de Wet for valuable comments about the manuscript.
ACKNOWLEDGMENTS
- 1. Temple IK, James RS, Crolla JA, Sitch FL, Jacobs PA, Howell WM, Betts P, Baum JD, Shield JP. An imprinted gene(s) for diabetes? Nat Genet 1995;9:110-112. ArticlePubMedPDF
- 2. Flanagan SE, Patch AM, Mackay DJ, Edghill EL, Gloyn AL, Robinson D, Shield JP, Temple K, Ellard S, Hattersley AT. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007;56:1930-1937. ArticlePubMedPDF
- 3. Gloyn AL, Reimann F, Girard C, Edghill EL, Proks P, Pearson ER, Temple IK, Mackay DJ, Shield JP, Freedenberg D, Noyes K, Ellard S, Ashcroft FM, Gribble FM, Hattersley AT. Relapsing diabetes can result from moderately activating mutations in KCNJ11. Hum Mol Genet 2005;14:925-934. ArticlePubMed
- 4. Greeley SA, Tucker SE, Worrell HI, Skowron KB, Bell GI, Philipson LH. Update in neonatal diabetes. Curr Opin Endocrinol Diabetes Obes 2010;17:13-19. ArticlePubMed
- 5. Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 2003;81:133-176. ArticlePubMed
- 6. Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 1989;54:87-143. PubMed
- 7. Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 2005;115:2047-2058. ArticlePubMedPMC
- 8. Inagaki N, Gonoi T, Clement JP 4th, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 1995;270:1166-1170. ArticlePubMed
- 9. Sakura H, Ammala C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscle. FEBS Lett 1995;377:338-344. PubMed
- 10. Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 1997;387:179-183. ArticlePubMedPDF
- 11. Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP 4th, Boyd AE 3rd, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 1995;268:423-426. ArticlePubMed
- 12. Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in KATP channel activation by Mg-ADP and diazoxide. EMBO J 1997;16:1145-1152. ArticlePubMedPMC
- 13. Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP 4th, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 1996;272:1785-1787. ArticlePubMed
- 14. Aittoniemi J, Fotinou C, Craig TJ, de Wet H, Proks P, Ashcroft FM. Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos Trans R Soc Lond B Biol Sci 2009;364:257-267. PubMed
- 15. Proks P, Arnold AL, Bruining J, Girard C, Flanagan SE, Larkin B, Colclough K, Hattersley AT, Ashcroft FM, Ellard S. A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum Mol Genet 2006;15:1793-1800. ArticlePubMed
- 16. Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 2006;355:456-466. ArticlePubMed
- 17. Patch AM, Flanagan SE, Boustred C, Hattersley AT, Ellard S. Mutations in the ABCC8 gene encoding the SUR1 subunit of the KATP channel cause transient neonatal diabetes, permanent neonatal diabetes or permanent diabetes diagnosed outside the neonatal period. Diabetes Obes Metab 2007;9(Suppl 2):28-39. ArticlePubMed
- 18. Ellard S, Flanagan SE, Girard CA, Patch AM, Harries LW, Parrish A, Edghill EL, Mackay DJ, Proks P, Shimomura K, Haberland H, Carson DJ, Shield JP, Hattersley AT, Ashcroft FM. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet 2007;81:375-382. ArticlePubMedPMC
- 19. Flanagan SE, Clauin S, Bellanne-Chantelot C, de Lonlay P, Harries LW, Gloyn AL, Ellard S. Update of mutations in the genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat 2009;30:170-180. ArticlePubMed
- 20. Zwaveling-Soonawala N, Hagebeuk EE, Slingerland AS, Ris-Stalpers C, Vulsma T, van Trotsenburg AS. Successful transfer to sulfonylurea therapy in an infant with developmental delay, epilepsy and neonatal diabetes (DEND) syndrome and a novel ABCC8 gene mutation. Diabetologia 2011;54:469-471. ArticlePubMed
- 21. Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, MacDonald MJ, Stoy J, Steiner DF, Philipson LH, Bell GI, Hattersley AT, Ellard S. Neonatal Diabetes International Collaborative Group. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008;57:1034-1042. PubMed
- 22. Edghill EL, Flanagan SE, Ellard S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev Endocr Metab Disord 2010;11:193-198. ArticlePubMedPDF
- 23. Fatehi M, Raja M, Carter C, Soliman D, Holt A, Light PE. The ATP-sensitive K+ channel ABCC8 S1369A type 2 diabetes risk variant increases MgATPase activity. Diabetes 2012;61:241-249. ArticlePubMedPDF
- 24. Lin YW, Akrouh A, Hsu Y, Hughes N, Nichols CG, De Leon DD. Compound heterozygous mutations in the SUR1 (ABCC 8) subunit of pancreatic KATP channels cause neonatal diabetes by perturbing the coupling between Kir6.2 and SUR1 subunits. Channels (Austin) 2012;6:133-138. ArticlePubMedPMC
- 25. Proks P, Ashcroft FM. Modeling KATP channel gating and its regulation. Prog Biophys Mol Biol 2009;99:7-19. ArticlePubMed
- 26. Proks P, Shimomura K, Craig TJ, Girard CA, Ashcroft FM. Mechanism of action of a sulphonylurea receptor SUR1 mutation (F132L) that causes DEND syndrome. Hum Mol Genet 2007;16:2011-2019. ArticlePubMed
- 27. Babenko AP, Vaxillaire M. Mechanism of KATP hyperactivity and sulfonylurea tolerance due to a diabetogenic mutation in L0 helix of sulfonylurea receptor 1 (ABCC8). FEBS Lett 2011;585:3555-3559. ArticlePubMedPMC
- 28. Zhou Q, Garin I, Castano L, Argente J, Munoz-Calvo MT, Perez de Nanclares G, Shyng SL. Neonatal diabetes caused by mutations in sulfonylurea receptor 1: interplay between expression and Mg-nucleotide gating defects of ATP-sensitive potassium channels. J Clin Endocrinol Metab 2010;95:E473-E478. ArticlePubMedPMC
- 29. de Wet H, Rees MG, Shimomura K, Aittoniemi J, Patch AM, Flanagan SE, Ellard S, Hattersley AT, Sansom MS, Ashcroft FM. Increased ATPase activity produced by mutations at arginine-1380 in nucleotide-binding domain 2 of ABCC8 causes neonatal diabetes. Proc Natl Acad Sci U S A 2007;104:18988-18992. ArticlePubMedPMC
- 30. Mannikko R, Flanagan SE, Sim X, Segal D, Hussain K, Ellard S, Hattersley AT, Ashcroft FM. Mutations of the same conserved glutamate residue in NBD2 of the sulfonylurea receptor 1 subunit of the KATP channel can result in either hyperinsulinism or neonatal diabetes. Diabetes 2011;60:1813-1822. ArticlePubMedPMCPDF
- 31. Ortiz D, Voyvodic P, Gossack L, Quast U, Bryan J. Two neonatal diabetes mutations on transmembrane helix 15 of SUR1 increase affinity for ATP and ADP at nucleotide binding domain 2. J Biol Chem 2012;287:17985-17995. ArticlePubMedPMC
- 32. de Wet H, Proks P, Lafond M, Aittoniemi J, Sansom MS, Flanagan SE, Pearson ER, Hattersley AT, Ashcroft FM. A mutation (R826W) in nucleotide-binding domain 1 of ABCC8 reduces ATPase activity and causes transient neonatal diabetes. EMBO Rep 2008;9:648-654. ArticlePubMedPMCPDF
- 33. Masia R, De Leon DD, MacMullen C, McKnight H, Stanley CA, Nichols CG. A mutation in the TMD0-L0 region of sulfonylurea receptor-1 (L225P) causes permanent neonatal diabetes mellitus (PNDM). Diabetes 2007;56:1357-1362. ArticlePubMedPDF
- 34. Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J Physiol 1997;504(Pt 1):35-45. ArticlePubMedPMC
- 35. Abdelmoneim AS, Hasenbank SE, Seubert JM, Brocks DR, Light PE, Simpson SH. Variations in tissue selectivity amongst insulin secretagogues: a systematic review. Diabetes Obes Metab 2012;14:130-138. ArticlePubMed
- 36. Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002;51(Suppl 3):S368-S376. ArticlePubMedPDF
- 37. Shield JP, Flanagan SE, Mackay DJ, Harries LW, Proks P, Girard C, Ashcroft FM, Temple IK, Ellard S. Mosaic paternal uniparental isodisomy and an ABCC8 gene mutation in a patient with permanent neonatal diabetes and hemihypertrophy. Diabetes 2008;57:255-258. ArticlePubMedPDF
- 38. Hambrock A, Loffler-Walz C, Quast U. Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br J Pharmacol 2002;136:995-1004. ArticlePubMedPMCPDF
- 39. Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT. Neonatal Diabetes International Collaborative Group. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 2008;31:204-209. ArticlePubMedPDF
- 40. Klupa T, Kowalska I, Wyka K, Skupien J, Patch AM, Flanagan SE, Noczynska A, Arciszewska M, Ellard S, Hattersley AT, Sieradzki J, Mlynarski W, Malecki MT. Mutations in the ABCC8 (SUR1 subunit of the KATP channel) gene are associated with a variable clinical phenotype. Clin Endocrinol (Oxf) 2009;71:358-362. ArticlePubMed
- 41. Vaxillaire M, Dechaume A, Busiah K, Cave H, Pereira S, Scharfmann R, de Nanclares GP, Castano L, Froguel P, Polak M. SUR1-Neonatal Diabetes Study Group. New ABCC8 mutations in relapsing neonatal diabetes and clinical features. Diabetes 2007;56:1737-1741. ArticlePubMedPDF
- 42. Stanik J, Gasperikova D, Paskova M, Barak L, Javorkova J, Jancova E, Ciljakova M, Hlava P, Michalek J, Flanagan SE, Pearson E, Hattersley AT, Ellard S, Klimes I. Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. J Clin Endocrinol Metab 2007;92:1276-1282. ArticlePubMedPDF
- 43. Codner E, Flanagan SE, Ugarte F, Garcia H, Vidal T, Ellard S, Hattersley AT. Sulfonylurea treatment in young children with neonatal diabetes: dealing with hyperglycemia, hypoglycemia, and sick days. Diabetes Care 2007;30:e28-e29. PubMed
- 44. Pearson ER, Flechtner I, Njolstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, Slingerland AS, Shield J, Robert JJ, Holst JJ, Clark PM, Ellard S, Sovik O, Polak M, Hattersley AT. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 2006;355:467-477. ArticlePubMed
- 45. Remedi MS, Kurata HT, Scott A, Wunderlich FT, Rother E, Kleinridders A, Tong A, Bruning JC, Koster JC, Nichols CG. Secondary consequences of beta cell inexcitability: identification and prevention in a murine model of KATP-induced neonatal diabetes mellitus. Cell Metab 2009;9:140-151. PubMedPMC
- 46. Kumaraguru J, Flanagan SE, Greeley SA, Nuboer R, Stoy J, Philipson LH, Hattersley AT, Rubio-Cabezas O. Tooth discoloration in patients with neonatal diabetes after transfer onto glibenclamide: a previously unreported side effect. Diabetes Care 2009;32:1428-1430. PubMedPMC
- 47. Shah B, Breidbart E, Pawelczak M, Lam L, Kessler M, Franklin B. Improved long-term glucose control in neonatal diabetes mellitus after early sulfonylurea allergy. J Pediatr Endocrinol Metab 2012;25:353-356. ArticlePubMed
- 48. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837-853. ArticlePubMed
- 49. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993;329:977-986. ArticlePubMed
REFERENCES
Fig. 1Stimulus-secretion coupling in pancreatic β-cells. (A) When extracellular glucose, and thus pancreatic β-cell metabolism, is low, adenosine triphosphate (ATP)-sensitive potassium (KATP) channels are open. As a result, the cell membrane is hyperpolarised. This keeps voltage-gated Ca2+ channels closed, so that Ca2+ influx remains low and no insulin is released. (B) When extracellular glucose concentration rises, glucose is taken up by the β-cell and metabolised. Metabolism generates ATP at the expense of magnesium adenosine diphosphate (MgADP), thereby closing KATP channels. This causes membrane depolarization, opening of voltage-gated Ca2+ channels, Ca2+ influx and insulin secretion.
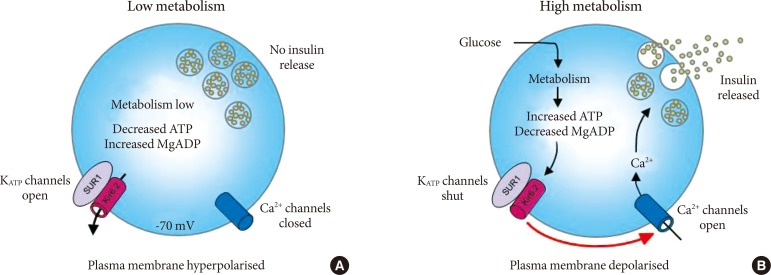
Fig. 2Location of neonatal diabetes mutations in the sulphonylurea receptor. Membrane topology of the sulphonylurea receptor with schematic representation of mutations which cause neonatal diabetes. Mutations showed in red and orange represent neonatal diabetes with developmental delay and epilepsy (DEND) and intermediate DEND syndrome respectively and grey colored mutations in italics transient neonatal diabetes; the rest of the mutations cause permanent neonatal diabetes. TMD, transmembrane domain; NBD, nucleotide binding domain.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Long-term Follow-up of Glycemic and Neurological Outcomes in an International Series of Patients With Sulfonylurea-Treated ABCC8 Permanent Neonatal Diabetes
Pamela Bowman, Frances Mathews, Fabrizio Barbetti, Maggie H. Shepherd, Janine Sanchez, Barbara Piccini, Jacques Beltrand, Lisa R. Letourneau-Freiberg, Michel Polak, Siri Atma W. Greeley, Eamon Rawlins, Tarig Babiker, Nicholas J. Thomas, Elisa De Franco, S
Diabetes Care.2021; 44(1): 35. CrossRef - Structure based analysis of KATP channel with a DEND syndrome mutation in murine skeletal muscle
Shoichiro Horita, Tomoyuki Ono, Saul Gonzalez-Resines, Yuko Ono, Megumi Yamachi, Songji Zhao, Carmen Domene, Yuko Maejima, Kenju Shimomura
Scientific Reports.2021;[Epub] CrossRef - Clinical and Genetic Characteristics of ABCC8 Nonneonatal Diabetes Mellitus: A Systematic Review
Meng Li, Xueyao Han, Linong Ji, Karim Gariani
Journal of Diabetes Research.2021; 2021: 1. CrossRef - Spacial models of malfunctioned protein complexes help to elucidate signal transduction critical for insulin release
Katarzyna Walczewska-Szewc, Wieslaw Nowak
Biosystems.2019; 177: 48. CrossRef - Cantu syndrome–associated SUR2 (ABCC9) mutations in distinct structural domains result in KATP channel gain-of-function by differential mechanisms
Conor McClenaghan, Alex Hanson, Monica Sala-Rabanal, Helen I. Roessler, Dragana Josifova, Dorothy K. Grange, Gijs van Haaften, Colin G. Nichols
Journal of Biological Chemistry.2018; 293(6): 2041. CrossRef - Hyperinsulinism-Causing Mutations Cause Multiple Molecular Defects in SUR1 NBD1
Claudia P. Alvarez, Marijana Stagljar, D. Ranjith Muhandiram, Voula Kanelis
Biochemistry.2017; 56(18): 2400. CrossRef - KATP Channel Mutations and Neonatal Diabetes
Kenju Shimomura, Yuko Maejima
Internal Medicine.2017; 56(18): 2387. CrossRef - Molecular action of sulphonylureas on KATP channels: a real partnership between drugs and nucleotides
Heidi de Wet, Peter Proks
Biochemical Society Transactions.2015; 43(5): 901. CrossRef - Successful development and use of a thermodynamic stability screen for optimizing the yield of nucleotide binding domains
Elvin D. de Araujo, Voula Kanelis
Protein Expression and Purification.2014; 103: 38. CrossRef - Reclasificación y transferencia de insulina a sulfonilureas en un paciente con mutación en KCNJ11 tras 15 años de tratamiento con insulina
Magdalena Capponi, Carmen Quirós, Ignacio Conget, Enric Esmatjes, Marga Giménez
Avances en Diabetología.2014; 30(4): 115. CrossRef
 PubReader
PubReader Cite
Cite