New Drugs for Treating Dyslipidemia: Beyond Statins
Article information
Abstract
Statins have been shown to be very effective and safe in numerous randomized clinical trials, and became the implacable first-line treatment against atherogenic dyslipidemia. However, even with optimal statin treatment, 60% to 80% of residual cardiovascular risk still exists. The patients with familial hypercholesterolemia which results in extremely high level of low density lipoprotein cholesterol (LDL-C) level and the patients who are intolerant or unresponsive to statins are the other hurdles of statin treatment. Recently, new classes of lipid-lowering drugs have been developed and some of them are available for the clinical practice. The pro-protein convertase subtilisin/kexintype 9 (PCSK9) inhibitor increases the expression of low density lipoprotein (LDL) receptor in hepatocytes by enhancing LDL receptor recycling. The microsomal triglyceride transport protein (MTP) inhibitor and antisense oligonucleotide against apolipoprotein B (ApoB) reduce the ApoB containing lipoprotein by blocking the hepatic very low density lipoprotein synthesis pathway. The apolipoprotein A1 (ApoA1) mimetics pursuing the beneficial effect of high density lipoprotein cholesterol and can reverse the course of atherosclerosis. ApoA1 mimetics had many controversial clinical data and need more validation in humans. The PCSK9 inhibitor recently showed promising results of significant LDL-C lowering in familial hypercholesterolemia (FH) patients from the long-term phase III trials. The MTP inhibitor and antisesnse oligonucleotide against ApoB were approved for the treatment of homozygous FH but still needs more consolidated evidences about hepatic safety such as hepatosteatosis. We would discuss the benefits and concerns of these new lipid-lowering drugs anticipating additional benefits beyond statin treatment.
INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of death and major health care burden in worldwide regardless of different ethnicities. Dyslipidemia, characterized by high levels of atherogenic lipoproteins including low density lipoprotein cholesterol (LDL-C), is known as the major risk factor of ASCVD [1]. Statins, the 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor, efficiently block hepatic cholesterol synthesis and lower LDL-C sufficiently up to 50% from the baseline according to statin potency [2]. During the past few decades, statins have been the corner stone of the medical treatment of dyslipidemia. Statins reduce ASCVD risk by 15% to 37% (Fig. 1), but residual 60% to 80% of ASCVD risk still remains [3]. These remaining ASCVD risk has been considered as causing the major vascular event in about 20% of patients with coronary heart disease even under the optimal statin treatment [4].
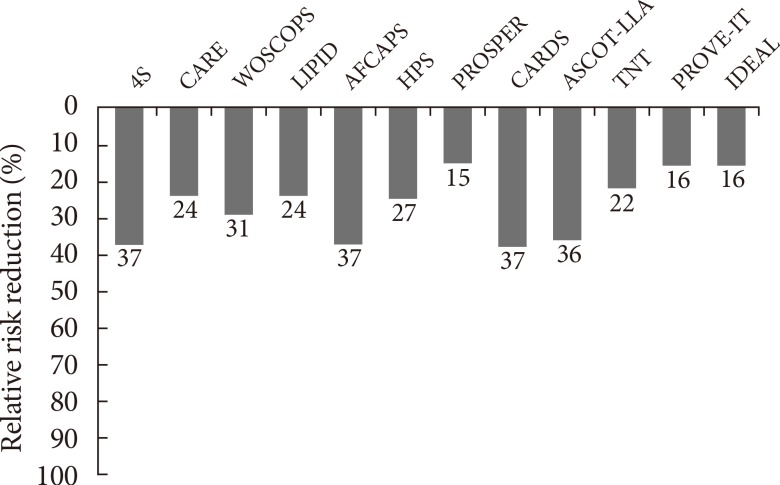
Residual risk for developing cardiovascular disease still remains despite proper range of low density lipoprotein cholesterol reduction: Results from many different statin trials. 4S, Scandinavian Simvastatin Survival Study; CARE, Cholesterol and Recurrent Events; WOSCOPS, West of Scotland Coronary Prevention Study; LIPID, Long-term Intervention with Pravastatin in Ischemic Disease; AFCAPS, Air Force/Texas Coronary Atherosclerosis Prevention Study; HPS, Heart Protection Study; PROSPER, Prospective Study of Pravastatin Elderly at Risk; CARDS, Collaborative Atorvastatin Diabetes Study; ASCOT-LLA, Anglo-Scandinavian Cardiac Outcomes Trial-Lipid-lowering Arm; TNT, Treating to New Targets; PROVE-IT, Pravastatin or Atorvastatin Evaluation and Infection Therapy; IDEAL, Incremental Decrease in End Points through Aggressive Lipid Lowering. Adapted from Lim et al., with permission from Elsevier [1].
Familial hypercholesterolemia is a genetic disorder caused by a mutation in low density lipoprotein (LDL) receptor (LDLR) gene, apolipoprotein B (ApoB) gene or pro-protein convertase subtilisin/kexintype 9 (PCSK9) gene with the prevalence of 1 in 300 to 500 people for heterozygous form and 1 in 1,000,000 people for the more severe homozygous form [5]. These genetic defects cause the significant elevation of blood LDL-C levels, which result in the early development of ASCVD and in higher mortality [5]. High dose statins are the first choice of treatment for these patients, but even with maximal intensity of statin treatment only 20% of patients with familial hypercholesterolemia achieve optimal LDL-C goal [5]. Furthermore, a subset of patients is intolerant to high dose statin therapy due to adverse effects including myotoxicity or hepatotoxicity.
Bile acid-binding resins, fibrates, niacin, and ezetimibe has been approved as non-statin agents for treating dyslipidemia [6]. Each class of non-statin drugs showed meaningful improvement of lipid profiles and especially has distinct effect in subtractions of blood lipoprotein composition such as elevating high density lipoprotein cholesterol (HDL-C) particles. However, none of these agents showed additional risk reduction of ASCVD when it is adding to the statin treatment. Only ezetimibe showed significant decrease of cardiovascular events from the recent randomized clinical trial: IMPROVE-IT, comparing simvastatin monotherapy and simvastatin plus ezetimibe combination [7].
There have been consistent needs how we could optimize the treatment for patients with higher risk of ASCVD. Because there are still many percentage of patients exist to request new drug combination beyond statin treatment. In this review, we will discuss four newly developed drugs for treating dyslipidemia, PCSK9 inhibitor, microsomal triglyceride transport protein (MTP) inhibitor, apolipoprotein A1 (ApoA1) mimetics, and antisense oligonucleotide against ApoB including their mode of actions and the results of preclinical and clinical studies.
PCSK9 INHIBITORS
Mode of action
PCSK9 is a serine protease that plays a central role in cholesterol metabolism in the liver by enhancing the degradation of LDLRs [8]. LDLR can be recycled or degraded in the lysosomal process after internalization. Circulating PCSK9 binds to the LDLRs directing the LDLRs to the lysosome, enhancing their clearance in the hepatocyte for degradation, and preventing the recycling of LDLRs back to the cell surface after internalization [9]. By blocking PCSK9, PCSK9 inhibitors can reduce LDLRs degradation and increase the surface expression of the LDLRs, which in turn enhances LDLRs recycling and reduces the LDL-C level (Fig. 2) [10]. Several approaches to inhibit PCSK9 have been proposed, including monoclonal antibody, small interfering RNA, antisense oligonucleotide, and mimetic peptides (Table 1) [11]. Among them, the fully humanized monoclonal antibody against PCSK9 showed successful human data by far [11].
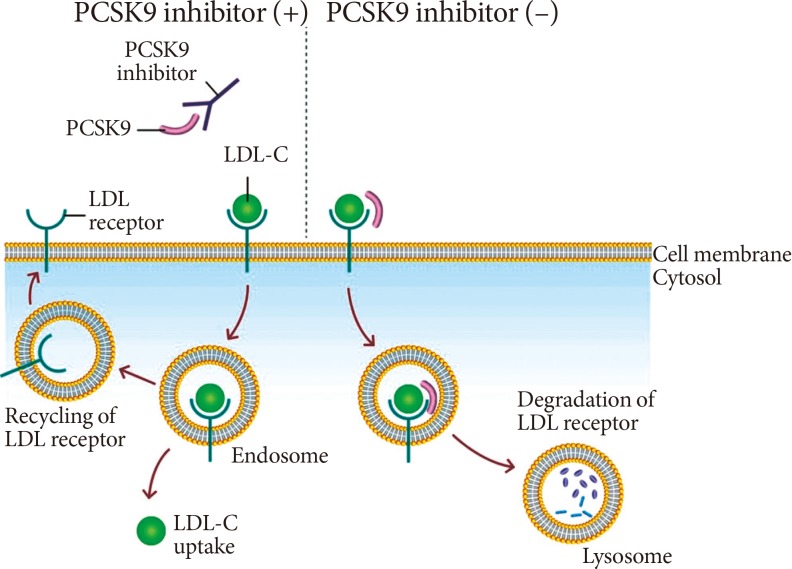
Therapeutic mechanism of pro-protein convertase subtilisin/kexin type 9 (PCSK9) inhibition. Binding of PCSK9 to the low density lipoprotein (LDL) receptor leads to the degradation of LDL receptor at lysosome. PCKS9 inhibitor, a monoclonal antibody against PCKS9, inhibits the binding of PCSK9 and LDL receptor, which results in the recycling of LDL receptor and increased expression of LDL receptor at cell membrane. LDL-C, low density lipoprotein cholesterol.
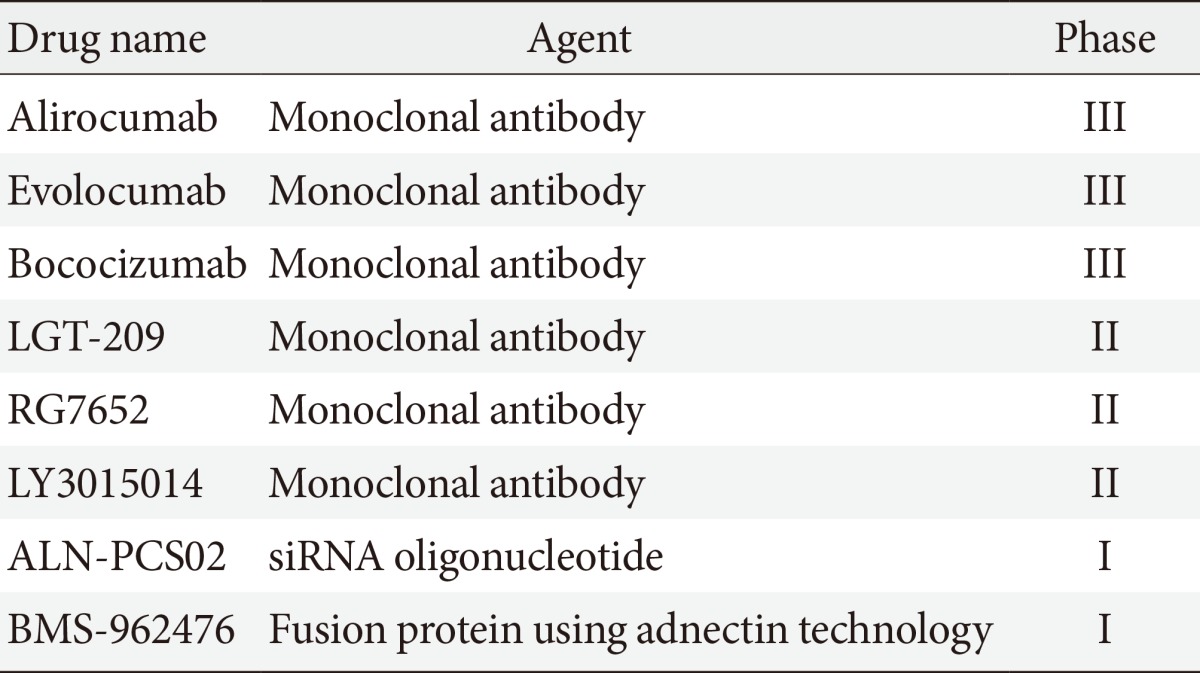
PCSK9-directed agents in development
Preclinical study
In mice with lacking PCSK9, the accumulation of cholesteryl esters in the lesion of aortic atherosclerosis was markedly reduced. By comparison, overexpression of PCSK9 induced an excess burden of atherosclerosis [12]. But in LDLR deficient mice, knock down or overexpression of PCSK9 had no significant effects on the cholesteryl ester accumulation and the size of atheromatous plaque. This study strongly suggests that the process by which PCSK9 enhances atherosclerosis is primarily mediated by its action on the LDLR [12]. Cloned guinea pigs created by transposition of a gain of function mutation of human PCSK9, a model for familial hypercholesterolemia, had a significant increase in aortic atherosclerosis compared with wild-type counterparts [13].
Clinical study
Among the various approaches to PCSK9 inhibition, the data for human studies are only available for monoclonal antibody against PCSK9. In phase II studies, the two most advanced monoclonal antibodies in development (alirocumab and evolocumab) decreased atherogenic lipoproteins very effectively and these drugs were well tolerated in human. In the clinical trial of 77 patients with heterozygous familial hypercholesterolemia, alirocumab reduced LDL-C by 29% to 43% for 150 to 300 mg injection at every 4 weeks and by 68% for 150 mg injection at every 2 weeks [14]. In addition, the higher dose of alirocumab, 150 mg every 2 weeks, showed significant increase in HDL-C and ApoA1 by 6.5% and 8.8%, respectively, as well as decrease in ApoB and non-HDL-C [14]. In the GAUSS trial which investigated the efficacy and safety of evolocumab in 160 statin-intolerant patients, evolocumab induced a significant dose-dependent decrease in LDL-C levels from 40.8% to 50.7% compared to baseline [15]. Furthermore, the combination of evolocumab and ezetimibe showed an almost 63% reduction in LDL-C. The result of a large phase III trial of alirocumab has been recently reported. In the trial involving 2,341 patients who were receiving maximum tolerated dose of statins, alirocumab reduced LDL-C by 62% compare to the baseline and the effect was maintained over 78 weeks of extended treatment [16]. Furthermore, in the post hoc analysis, alirocumab showed reduced rate of major adverse cardiovascular events compare to the placebo (hazard ratio, 0.52; 95% confidence interval, 0.31 to 0.90) [16]. The result of the long-term extension study of evolocumab trials also showed the consistent LDL-C reduction until 48 weeks of treatment and reduced cardiovascular events at approximately 1 year of treatment (hazard ratio, 0.47; 95% confidence interval, 0.28 to 0.78) [17].
MTP INHIBITOR
Mode of action
MTP is predominantly expressed in hepatocytes and enterocytes, whose action is required in the synthesis of ApoB-containing lipoproteins. MTP transfers triglyceride (TG), phospholipids and cholesteryl esters to the ApoB in endoplasmic reticulum and has a critical role in the synthesis of very low density lipoprotein (VLDL) and chylomicrons in liver and intestine [18,19]. Inhibition of MTP results in the decreased synthesis and secretion of VLDL in the liver by inhibiting the lipidation of ApoB (Fig. 3) [20]. MTP inhibition can reverse the increased hepatic production and secretion of VLDLs caused by insulin resistance. Moreover, inhibition of MTP in enterocytes can contribute to the reduction in plasma TG level by reducing dietary fat absorption through chylomicron. An orally active small molecule inhibitor of MTP, lomitapide, was developed and it has been approved for the treatment of homozygous familial hypercholesterolemia [21].
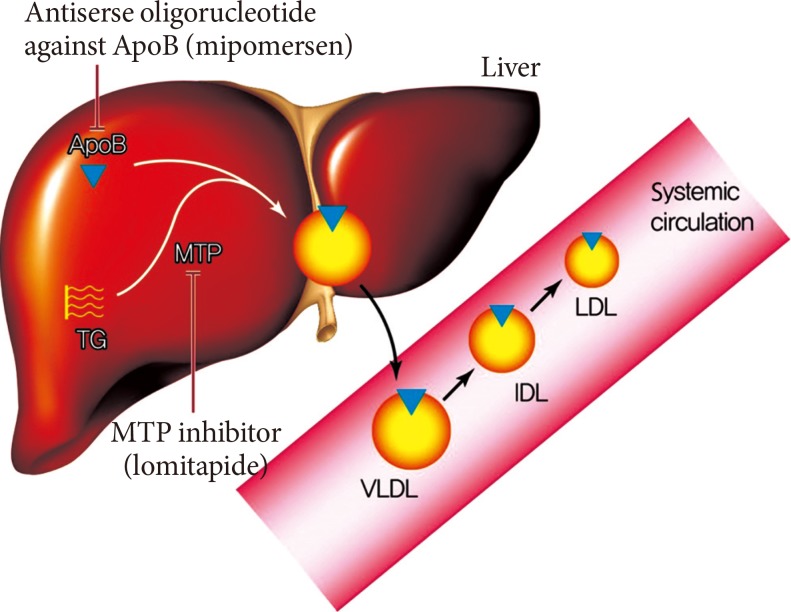
Therapeutic mechanism of lomitapide and mipomersen. The assembly of very low density lipoprotein (VLDL) requires the loading of triglyceride (TG) to the apolipoprotein B (ApoB) in the liver. The microsomal triglyceride transfer protein (MTP) works in this proccess and transfers TG to the ApoB. The secreted VLDL is converted to low density lipoprotein (LDL) in the bloodstream. The lomitapide inhibits the action of MTP and mipomersen inhibits the synthesis of ApoB. These two agents eventually inhibit the assembly of VLDL in the liver, which results in decreased LDL in the bloodstream. IDL, intermediate-density lipoprotein.
Preclinical study
In an early study, treatment of hamsters with lomitapide for 7 days resulted in a dose-dependent reduction in both VLDL and LDL-C in the range of 19% to 89% and TGs in the range of 8% to 49% [22]. However, a concomitant decrease in HDL-C levels, particularly at higher doses, was noticed. The same study also investigated the efficacy of lomitapide in homozygous Watanabe-heritable hyperlipidemic rabbits as an atypical model of homozygous familial hypercholesterolemia. After the administration of lomitapide for 2 weeks, the ApoB-containing lipoprotein level was normalized [22]. The single-dose administration of lomitapide to the Zucker fatty rat, a genetic model of diabetic dyslipidemia and metabolic syndrome, reduced the serum level of TG by 35% and 47% at 0.3 and 1 mg/kg doses. Longer duration of treatment also showed significant decrease in the serum level of TG (71% to 87%), nonesterified fatty acids (33% to 40%), and LDL-C (26% to 29%) [23].
Clinical study
In a phase III trial of 29 homozygous familial hypercholesterolemia with mean baseline LDL-C of 336 mg/dL despite the previous lipid-lowering therapy, lomitapide decreased LDL-C by 50% at 26 weeks of treatment and 38% at 52 weeks of treatment [24]. The HDL-C levels were significantly reduced by 12% at 26 weeks, but returned to the pretreatment levels at 78 weeks [24]. Common adverse effects include diarrhea, nausea, and abdominal pain [4]. In the early phase clinical studies, lomitapide increased hepatic TG content in a dose dependent manner, presumably due to hepatic MTP inhibition [6] and trapping VLDLs in the liver. In the phase III trial, 10 patients showed elevation of hepatic enzyme and resolved with dose modification. Results from this study showed acceptable risk-to-benefit profile and lomitapide was approved for the treatment for homozygous familial hypercholesterolemia by US Food and Drug Administration (FDA) [21].
ANTISENSE OLIGONUCLEOTIDE AGAINST APOLIPOPROTEIN B
Mode of action
ApoB is the major structural protein of atherogenic lipoproteins (APO-B containing lipoproteins). It has a key role in the assembly and secretion of VLDL from the liver [25]. Plasma ApoB concentration is a reliable index of the total number of atherogenic lipoproteins such as small dense LDL-C [25]. Mipomersen is a synthetic 20 nucleotide antisense oligonucleotide which can bind to ApoB mRNA via complementary sequence interactions [21]. Hybridization of mipomersen to the target ApoB mRNA creates a substrate for RNase H1, which results in the decrease of the ApoB mRNA level and the production of ApoB protein (Fig. 3) [21]. Two chemical modifications were made to the nucleotide structure of mipomersen. First, the internucleotide linkage was chemically modified as a phosphorothioated ester, which results in the resistance to the hydrolysis or degradation by nucleases and increase of binding to plasma protein to facilitate drug distribution and absorption. The second modification, insertion of methoxyethyl sugar residues into the first and last five positions made the mipomersen gave more stability and increased affinity [21,25].
Preclinical study
Including various animal studies such as mouse, hamster, rabbit, and monkey, the species-specific antisense oligonucleotide against ApoB reduced hepatic ApoB-100 mRNA and protein level, as well as serum levels of ApoB, LDL-C, and total cholesterol, in a dosedependent manner [21]. After subcutaneous injection, mipomersen is readily absorbed and distributed to tissues with highest drug concentrations in the liver and kidney. Mipomersen is metabolized by nucleases and the plasma elimination half-life ranges from 1 to 2 months, which allows relatively infrequent dosing. The clinical dosing regimen for mipomersen is weekly [21].
Clinical study
For the subjects with mild dyslipidemia, 12 weeks treatment with mipomersen with doses of 50 to 400 mg every 3 weeks resulted in a dose-dependent reduction in ApoB and LDL-C by a maximum of 50% and 35%, respectively [26]. The efficacy of mipomersen therapy in patients with familial hypercholesterolemia was confirmed in several phase II and III trials. The phase III trial in 44 heterozygous familial hypercholesterolemia patients showed significant reductions in ApoB and LDL-C with maximum reduction of 33% and 34%. In another phase III trial in homozygous familial hypercholesterolemia patients who is already receiving maximum tolerated dose of lipid-lowering therapy, 26 weeks treatment of mipomersen resulted in placebo adjusted reduction of ApoB and LDL-C by 24% and 21% [27]. From the result of this trial, mipomersen approved by US FDA for the treatment of homozygous familial hypercholesterolemia. Other clinical trials in patients with primary hypercholesterolemia also showed dose-dependent and consistent reduction of ApoB and LDL-C with treatment of mipomersen [28]. Frequent adverse effects associated with mipomersen are injection site reactions, flu-like symptoms and hepatic enzyme elevation. The major safety concern is increased hepatic accumulation of TG presumably due to impaired VLDLs secretion, which is very similar mechanism of TG trapping as in MTP inhibitor. The transaminase elevations are reversible with dose adjustment or even transient with continued treatment, and the hepatic fat increases occur early and are stable over time [21]. The long-term clinical consequences of increased hepatic TG are unknown. There is still concern for regular monitoring of hepatic function is requested in the patients receiving mipomersen [6].
APOLIPOPROTEIN A1 MIMETICS
Mode of action
The high serum level of HDL-C is a well-known protective factor of ASCVD [29]. ApoA1 is the major apolipoprotein component of mature HDL. ApoA1 took cholesterol from macrophages in atherosclerotic lesions via ATP-binding cassette A1 (ABCA1), triggering reverse cholesterol transport. The central role of ApoA1 in comprising HDL-C makes it as an attractive target for modifying ASCVD risk. ApoA1 mimetics are a class of drugs that is designed to mimic the effect of ApoA1 and HDL-C to reverse the progression of atherosclerosis [30].
Preclinical study
Several investigators have tested the effect of direct infusion of HDL or recombinant HDL with different ApoA1 preparations on atherosclerosis [31]. In the study of animal models, the infusion of HDL or recombinant HDL showed beneficial effect and even reversal of atherosclerosis, but they were difficult to be developed as a form of administrable drug [31].
In 1980, Franceschini et al. [32] presented an ApoA1 variant called ApoA1 Milano among three Italian individuals with long lifespans and low atherosclerotic burden, despite low HDL levels and increased TGs. This genetic variant has a characteristic arginine to cysteine substitution, which allows for the formation of ApoA1 dimers [33]. The recombinant ApoA1 Milano, ETC-216, was created by combining the mutant HDL with phospholipids to create an HDL-like particle. The efficacy of ETC-216 was tested in cholesterol-fed carotid artery-injured rabbits. After two treatments, the lower doses of ETC-216 led to reduced lesion progression and the higher doses led to lesion regression and a significant reduction in markers associated with plaque instability [34].
The preliminary success of ApoA1 Milano and recombinant HDL led to the development of a small ApoA1 mimetic peptide capable of oral administration without the need for weekly infusion therapy. This mimetic peptide, called D-4F, was synthesized from D-amino acids and was shown to decrease atherosclerotic lesion volume by 79% in LDLR-null mice, despite no change in plasma HDL [35]. Since its discovery, D-4F and its optical isomer L-4F have been shown to alter measures of cardiovascular disease in numerous animal models [36,37]. In vitro studies have shown that the mechanisms by which D-4F decreases atherosclerosis include increased cholesterol efflux from macrophages via ABCA1, increased transport of cholesterol to the liver via SR-B1, decreased monocyte chemotaxis and adhesion, and binding of oxidized lipids [38].
Clinical study
In a clinical study in patients with acute coronary syndrome, 5 weeks infusion of recombinant ApoA1 Milano decreased 4.2% of atheroma volume from baseline as measured by intravascular ultrasound [39]. Recombinant HDL containing normal human ApoA1 combined with phospholipid were also tested. In the ERASE (Effect of rHDL on Atherosclerosis Safety and Efficacy) study, patients with ACS received recombinant HDL (CSL-112) for 4 weeks, which resulted in no significant effect on atheroma or plaque volume compared with placebo [40]. However, compare to the baseline, the atheroma volume was significantly reduced by 3.4% [40]. In a phase I trial of small ApoA1 mimetic peptide, patients with coronary heart diseases received a single dose of D-4F, which resulted in a significantly improved HDL-inflammatory index relative to placebo [41]. L-4F showed the equal efficacy to D-4F when injected intravenously. However, Watson et al. [42] demonstrated that patients with CHD received intravenous L-4F over 7 days, showed no significant reduction in HDL-inflammatory index. Clearly, more preclinical and clinical studies including clinical trials of advanced phases are needed for ApoA1 mimetics. It is too early to make a conclusion on whether ApoA1 mimetics can be a clinically meaningful part of lipid-lowering treatment.
CONCLUSIONS
Statin therapy is a touchstone in the treatment of dyslipidemia. From numerous randomized clinical trials, it has been shown to be safe and efficacious for preventing future cardiovascular events. However, still, significant amount of residual ASCVD risk is remaining even under optimal statin treatment and significant portion of patients are intolerant or unresponsive to statin therapy. Many researchers and pharmaceutical companies are involved in this field of fighting for atherogenic dyslipidemia and it have been many promising results coming to apply in real clinical settings.
The PCSK9 inhibitor facilitates the uptake of LDL-C by enhancing LDLR recycling. It showed favorable effects for additional lowering of LDL-C when adding on to statin and nice safety profile with consistent long-term efficacy in large phase III trials. The MTP inhibitor and antisense oligonucleotide against ApoB are reducing ApoB-containing lipoprotein, the major atherogenic lipoprotein. Lomitapide, the MTP inhibitor, and mipomersen, the antisense oligonucleotides against ApoB, have shown their efficacy in lowering LDL-C in recent phase III trials and they were already approved for treating patients with homozygous familial hypercholesterolemia. Those two drugs are still in a major safety concern, which is increased hepatic fat accumulation as trapping TG due to their pharmacologic effect of inhibiting hepatic VLDL secretion. The long term safety profiles need to be evaluated in a near future. The ApoA1 mimetic is the most experimental class of drugs among four different classes in this review. It has been shown to alter or reverse the natural course of atherosclerosis despite the range of LDL-C level in preclinical studies. However, their efficacy seems to be modest and the results are not consistent from previous studies. It awaits further validation through various human studies.
The new classes of drugs beyond statin could enlighten the improvement for anti-atherosclerosis therapy. Clinicians should keep their eyes on the results of upcoming studies using new class of drugs to find the best and the optimal treatment modality for patients with dyslipidemia.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.