Immune-Checkpoint Inhibitors-Induced Type 1 Diabetes Mellitus: From Its Molecular Mechanisms to Clinical Practice
Article information

Abstract
With the increasing use of immune-checkpoint inhibitors (ICIs), such as anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and anti-programmed cell death-1 (PD-1), for the treatment of malignancies, cases of ICI-induced type 1 diabetes mellitus (ICI-T1DM) have been reported globally. This review focuses on the features and pathogenesis of this disease. T1DM is an immune-related adverse event that occurs following the administration of anti-PD-1 or anti-programmed death ligand-1 (PD-L1) alone or in combination with anti-CTLA-4. More than half of the reported cases presented as abrupt-onset diabetic ketoacidosis. The primary mechanism of ICI-T1DM is T-cell stimulation, which results from the loss of interaction between PD-1 and PD-L1 in pancreatic islet. The similarities and differences between ICI-T1DM and classical T1DM may provide insights into this disease entity. ICI-T1DM is a rare but often life-threatening medical emergency that healthcare professionals and patients need to be aware of. Early detection of and screening for this disease is imperative. At present, the only known treatment for ICI-T1DM is insulin injection. Further research into the mechanisms and risk factors associated with ICI-T1DM development may contribute to a better understanding of this disease entity and the identification of possible preventive strategies.
INTRODUCTION
Immunotherapy using immune-checkpoint inhibitors (ICIs) has improved cancer management and has become a cornerstone of cancer treatment over the past decade [1]. The successful use of ICIs has improved our understanding of the human immune system and cancer treatments. In 2011, ipilimumab, the first ICI targeting anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), was approved for use in cancer treatment [1]. Thereafter, several ICIs have been developed and authorized for use; the inhibitory immune checkpoints targeted by ICIs include CTLA-4, programmed cell death-1 (PD-1), and programmed death ligand-1 (PD-L1) [1,2]. In various clinical settings, ICIs are administered alone or in combination with other ICI classes or cytotoxic chemotherapy for 17 malignancies (Table 1) [2,3]. Recently, durable responses to these treatments have been reported, which led to their persistent use. Thus, characterization of the long-term physiological adverse effects of ICI treatment is important [4]. The mechanism of action of ICIs is mainly based on the inhibition of the physiological brake of immune activation; thus, they inevitably exert off-target effects that cause immune-related adverse events (irAEs) in various organs or tissues. This distinct entity of ICI-related adverse events prompts physicians and oncologists to provide immediate and appropriate treatment to patients developing irAEs due to the use of ICIs.

Currently available immune-checkpoint inhibitors
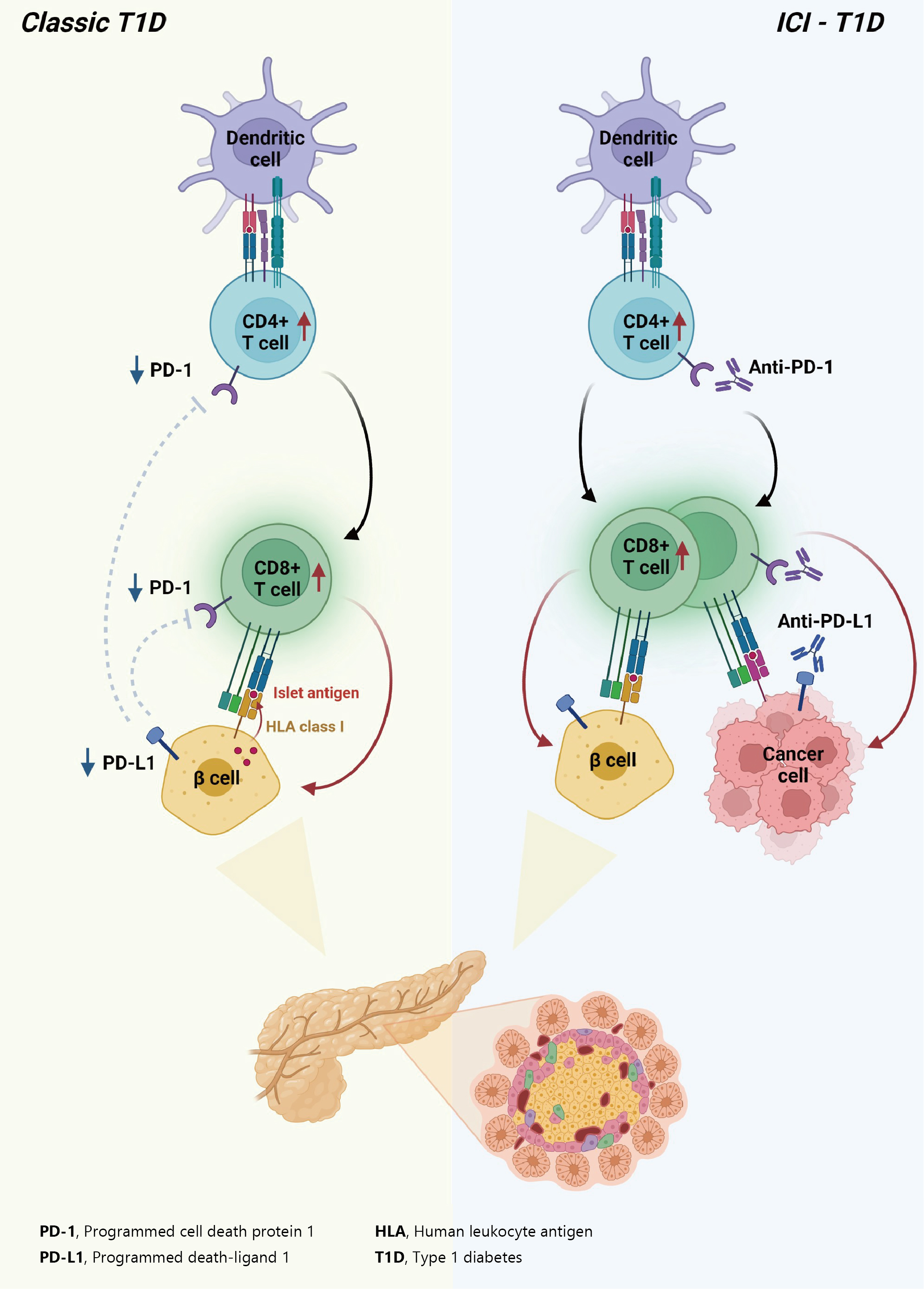
Immune checkpoints are receptors expressed by immune cells that enable dynamic control of immune homeostasis [5]. The attachment of PD-1 to PD-L1 on activated T-cells causes T-cell exhaustion, which is characterized by diminished effector function (cytotoxicity or cytokine production) and weak immunological responses to stimuli [6,7]. Tumor cells use this relationship to induce immune tolerance [8]. CTLA-4 is increased on the surface of activated T-cells to avoid overstimulation of T-cell receptors [9,10]. CTLA-4 competes with CD28, a T-cell receptor co-stimulatory receptor, for the binding between B7-1 and B7-2, which suppresses CD28-mediated T-cell activation [8,11]. The oncogenic and immunosuppressive nature of the tumor microenvironment is characterized by PD-L1 overexpression in cancer cells as well as PD-1 and CTLA-4 overexpression in T-cells [12]. Collectively, the inhibition of these molecules results in an immune-mediated antitumor response (Fig. 1) [11].
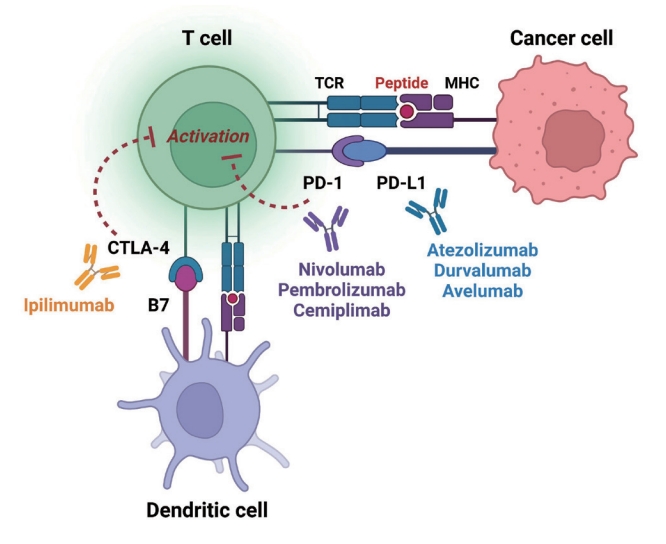
Effect of immune-checkpoint inhibitors on T lymphocytes. Anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA4) blocking and anti-programmed cell death-1 (PD-1) or antiprogrammed death ligand-1 (PD-L1) blocking restore pro-activatory signaling and result in effective antitumor T lymphocyte responses. TCR, T-cell receptor; MHC, major histocompatibility complex.
Immunological stimulation that exacerbates the severity of irAEs may be accompanied by an anticancer immune response. This specific ICI mechanism is supported by a small but reproducible positive connection between therapeutic responses and the incidence of irAEs [13-15]. Although the precise mechanisms underlying irAEs are unknown, they are assumed to be “bystander effects” of activated T-cells and are consistent with the mechanism of action of ICIs [13,16,17]. However, some non-antitumor pathways, such as those involving microbiota, viruses, or tissue-specific factors, are thought to cause irAEs [18-23]. Thus, “one-size-fits-all” mechanistic explanations become unlikely. The irAEs are most likely a result from factors that are or are not related to tumor. Chronic irAEs involving the endocrine system were the first to be reported, occurring in 15% to 40% of patients receiving ICIs [5]. Recently, ICI-induced type 1 diabetes mellitus (ICI-T1DM) has been proposed as a clinically significant and urgent irAE of ICIs. Herein, we aimed to review published articles on ICI-T1DM and experimental studies and discuss its mechanisms and pathogenesis as well as its distinct clinical characteristics.
PATHOGENESIS OF ICI-T1DM
Role of PD-1/PD-L1 in classical T1DM
T1DM is a chronic autoimmune disease triggered by both genetic and environmental factors, in which inappropriate hyperactivity in immune system causes destruction and dysfunction of insulin-producing β-cells [24,25]. Recently, teplizumab, a humanized anti-CD3 monoclonal antibody, was firstly approved for clinical use to delay the onset of T1DM, supporting the notion that T1DM is an immune disease and immunomodulation prior to clinical disease onset may be beneficial [26,27]. Human leukocyte antigen (HLA) antigens were first associated with insulin-dependent diabetes in 1973 [28], which established the relationship between the immune system and T1DM for the first time. Thereafter, genome-wide association studies have proven that HLA genes (particularly HLA class II loci) account for up to 50% of the genetic risk of T1DM, suggesting that the selective presentation of certain autoantigen peptides plays a role in T1DM pathogenesis [29-32]. Most T1DM cases are caused by immune-mediated death of pancreatic insulin-producing cells during an inflammatory process involving many immune cell types, including T lymphocytes [33]. Macrophages and dendritic cells enter the islets and migrate to the pancreatic lymph nodes, where they use antigen presentation to attract naive CD4+ T lymphocytes. These antigen-presenting cells (APCs) express β-cell antigens on major histocompatibility complex (MHC) class II molecules, causing CD4+ T lymphocytes to become activated. Th1 CD4+ T-cells interact with APCs, and the produced cytokines and free radicals drive CD8+ T-cell migration toward the islets. CD4+ and CD8+ T lymphocytes are the ultimate culprits of pancreatic β-cell death [34,35]. The immune system has been involved in the etiology of T1DM as immunotherapeutic therapies have demonstrated transient benefit in slowing down disease progression [36]. In other words, the T1DM response is analogous to effective anticancer immunity generated by immunotherapeutic suppression of PD-1 or its ligand, PD-L1, which otherwise controls autoimmune reactions. Other immunological and autoimmune responses, particularly those against pancreatic islets, may be generated by the use of ICIs in addition to anticancer immunity.
The protein PD-L1 exists in a variety of hematological and parenchymal cells, including pancreatic islet cells [37]. Transgenic overexpression of PD-L1 in pancreatic β-cells protected mice from developing diabetes; the comparison of PD-L1 transgenic mice with littermate controls led to the findings of reduced degree of insulitis, delayed disease onset, and dramatically reduced T1DM incidence [38]. The frequency of PD-L1+ cells and pancreatic surface expression of PD-L1 rapidly increased in nonobese diabetic (NOD) mice as they aged and developed diabetes, and infiltration of T-cells was significantly associated with increased cellular PD-L1 expression [39]. This could be explained by the fact that elevated PD-L1 expression in pancreatic β-cells is a protective mechanism designed to prevent the formation of self-reactive T-cells [39].
Experimental and clinical research has suggested that the alteration in PD-1/PD-L1 pathway is one of major pathogenesis in the development of autoimmune T1DM [34]. Ansari et al. [40] found that PD-1 or PD-L1 blocking caused diabetes in pre-diabetic NOD mice, whereas CTLA-4 inhibition caused diabetes only in neonates. Pauken et al. [41] demonstrated that in the absence of PD-1, CD4+ cells penetrated deeply into the islet core, which transformed peri-insulitis into damaging insulitis. This study suggested a concept wherein PD-1 regulates islet-reactive CD4+ T-cells in a cell-intrinsic manner by restricting proliferation and limiting pancreatic infiltration, and eventually prevents diabetes [41].
Tsutsumi et al. [42] conducted the first clinical study demonstrating that people with T1DM exhibited low PD-1 expression levels on activated T-cells. The researchers observed that in T1DM patients, the PD-1 expression levels in the peripheral CD4+ T-cells were significantly lower than those of healthy controls, indicating that decreased PD-1 gene expression in CD4+ T-cells may be related to the development of classic T1DM [42]. Contrarily, despite previous reports of elevated blood CTLA-4 levels in autoimmune disorders, the serum CTLA-4 levels did not significantly differ between T1DM patients and non-diabetic teenagers [43]. Colli et al. [44] employed immunofluorescence to examine the expression of PD-L1 in T1DM donor samples and found that PD-L1 was present in both young and older individuals with T1DM; however, it was absent in insulin-deficient islets where β-cells had been killed. Interestingly, a Chinese cohort study found that the partial remission (or honeymoon) phase of T1DM is accompanied by restoration of the PD-1/PD-L1 expression in peripheral CD4+ and CD8+ T-cells. These data indicate that the PD-1/PD-L1 pathway could be a viable target for therapeutics aimed at extending this stage, highlighting the significance of this pathway in T1DM etiology [45]. As a result, it can be hypothesized that the decreased PD-1 expression of T-cells stimulates autoreactive T-cells that infiltrate pancreatic islet cells, leading to the development of T1DM [34,46].
ICI-T1DM: proposed pathogenesis
Consistent with the proposed association between PD-1/PD-L1 and T1DM, ICIs which aimed to enhance antitumor immunity by inhibiting PD-1/PD-L1 pathway could result in the unintentional adverse effect including T1DM, due to the loss of indispensible immune regulation [47,48]. In theory, PD-1 inhibition through PD-1 or PD-L1 pharmacological inhibition increases pancreatic β-cell infiltration and death through activated autoreactive T-cells [24]. A recent study demonstrated that activated autoreactive T-cells respond to PD-1 inhibition by releasing interferons (IFNs), which activate monocyte-derived macrophages [49]. These T-cells use nitric oxide to kill pancreatic β-cells, which leads to insulin deficiency and ICI-T1DM. An examination of a patient’s pancreatic pathology revealed an increase in CD8+ T-cells relative to CD4+ T-cells as well as the absence of macrophages [50]. Another study found evidence of pancreatic inflammation, including pancreatic shrinkage, increased pancreatic enzyme levels, and peri-islet lymphocytic infiltration, in a patient who died with ICI-T1DM [51]. In the same study, anti-PD-L1, but not anti-CTLA-4, caused rapid development of diabetes in the NOD mouse model [51]. When treated with anti-PD-L1, cytolytic IFN-γ+CD8+ T-cells infiltrated islets and IFN-γ and tumor necrosis factor α (TNF-α) induced transcriptional changes suggesting dedifferentiation in pancreatic β-cells [51]. In vitro, IFN increases checkpoint ligand expression and activates apoptotic pathways in human β-cells, and in NOD mice, treatment with anti-PD-L1, anti-IFN, and anti-TNF prevented ICI-T1DM [51].
Clinically, T1DM does not develop in all patients receiving ICI treatment; only a very small proportion, approximately 1% of patients, develop ICI-T1DM. Additionally, the ICI-T1DM occurs several weeks to months after the initiation of ICI, suggesting that this immune-related type of diabetes may require sequential events. The events required for ICI-T1DM are not yet established, but the two-hit hypothesis, which involves an increase in PD-L1 expression in stressed β-cells and PD-L1/PD-1 blockade, is a possible explanation [52]. Notably, in NOD mice, PD-L1 expression in β-cells correlated with increased age and immune infiltrate in the islets, with diabetic mice displaying the highest PD-L1 levels [39,53]. Moreover, an adaptive immune response within the islets seems to be critical for the up-regulation of PD-L1 [39,53]. In the human pancreas, PD-L1 expression in islets was present in autoantibody-positive T1DM patients at greater levels than type 2 diabetes mellitus (T2DM) patients and healthy controls [39]. Thus, an increase in PD-L1 expression in the pancreas is likely indicative of an attempt to subdue an inflammatory response, which could be the first step in developing ICI-T1DM. Following the increase in PD-L1, particularly in certain patients, exposure to ICI may be the second and final trigger for the development of T1DM.
In summary, ICIs that target the PD-1/PD-L1 pathway induce transcriptional changes in cells and immunological infiltrates, which may lead to the development of diabetes [51]. Other immunological and autoimmune responses, particularly those against pancreatic islets, may be generated by the use of ICIs in addition to anticancer immunity. Fig. 2 depicts the similarities and differences between classic T1DM and ICI-T1DM.
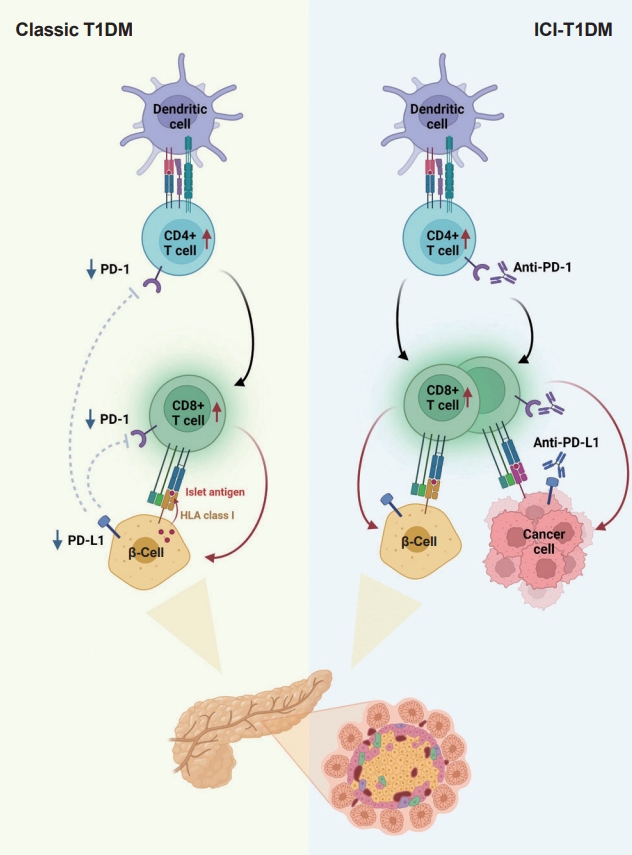
Pathogenesis of classic type 1 diabetes mellitus (T1DM) and immune-checkpoint inhibitor-induced T1DM (ICI-T1DM). In T1DM, inappropriate immune reaction can lead to an autoimmune response by autoreactive T-cells. The programmed cell death-1 (PD-1)/programmed death ligand-1 (PD-L1) inhibitory pathway plays a fundamental role in the maintenance of immune tolerance, thus, dysregulation of the PD-1/PD-L1 is an important pathogenesis of classical T1DM. Immunotherapeutic inhibition of PD-1 or its receptor PD-L1 to target cancer cells, involves autoimmune reactions which resembles the pathogenesis of T1DM. With immune-checkpoint inhibitors, unintended immune and autoimmune responses including those against pancreatic islets might be activated. HLA, human leukocyte antigen.
EPIDEMIOLOGY, CLINICAL CHARACTERISTICS, AND CURRENT GUIDELINES OF ICI-T1DM
Epidemiology of ICI-T1DM
In the past decade, the number of reported ICI-T1DM cases has consistently and significantly increased [47,54-60]. Generally, the incidence rate of T1DM following ICI administration is expected to be 1% [59,61,62]. In detail, the incidence rates of T1DM caused by nivolumab (anti-PD-1), pembrolizumab (anti-PD-1), durvalumab (anti-PD-L1), and atezolizumab (anti-PD-L1) were reported to be 3.5%, 2.3%, 0.3%, and 0.7%, respectively [63]. Among ICIs, anti-PD-1 and anti-PD-L1 are the most possible causes of T1DM. Although anti-CTLA-4 antibody monotherapy is rarely associated with T1DM development, the combination of anti-CTLA-4 with those against PD-1 or PD-L1 increases the risk of ICI-T1DM [58].
Clinical characteristics of ICI-T1DM from previous reports
Based on previous reports, we summarized some clinical features of ICI-T1DM. The most common agent that causes ICI-T1DM is anti-PD-1, and rapid β-cell destruction and insulin deficiency occur as early as 5 days following ICI administration and up to several months following ICI discontinuation [59,64]. Median time to diagnosis of ICI-T1DM found to vary significantly across different studies, ranging from 7 to 25 weeks [52, 65,66]. The rapid onset of this disease causes fulminant presentation, including diabetic ketoacidosis (DKA), in 40% to 76% of the affected patients [47,59,64,65,67-69]. Majority of the patients report overt insulin insufficiency and low C-peptide levels (0.3 ng/mL in 63.4% of patients) [54]. Anti-glutamic acid decarboxylase autoantibodies were detected in 43% (positive total islet autoantibody was observed in 20% to 71% of patients) of patients and increased pancreatic enzyme in some [54]. In most cases, insulin deficiency is irreversible; thus, majority of the patients eventually need lifelong insulin therapy [47,59,67]. Limited information is available regarding ethnic disparities in the incidence of ICI-T1DM. A review published in 2019 including a total of 90 cases with ICI-T1DM reported that individuals of Asian ethnicity accounted for 15% of the cases [48]. A more recent cohort study which will be elaborated on in the following paragraph, revealed that race/ethnicity (categorized as White, Black, Asian, and Hispanic) did not significantly affect the risk of developing ICI-T1DM [58]. The patients’ clinical features are summarized in Table 2.

Clinical features from previous studies and risk factors of immune-checkpoint inhibitor-induced type 1 diabetes mellitus
Recently, a study on the incidence and characteristics of ICI-T1DM in a large de-identified cohort of patients was published in the United States [58]. The authors investigated patient/treatment factors related to the incidence of ICI-T1DM, which developed in 261 (0.86%) of 30,337 patients. The combined use of anti-CTLA-4 with anti-PD-1 or anti-PD-L1 (hazard ratio [HR], 1.62; 95% confidence interval [CI], 1.15 to 2.26), young age (HR, 1.19 for every 5-year drop; 95% CI, 1.13 to 1.25), and preexisting diabetes (HR, 4.48; 95% CI, 3.45 to 5.83) were related to an increased risk of ICI-T1DM [58]. Although the risk of ICI-T1DM is low (0.86%), over 30% of patients with ICI-T1DM present with major acute complications, such as DKA or pancreatitis. Therefore, clinicians should educate high-risk patients regarding the dangers of ICI-T1DM and symptoms of hyperglycemia, such as polydipsia and polyuria. Early detection of and intervention for ICI-T1DM, such as glucose level monitoring and insulin administration, are extremely important.
A recent longitudinal trajectory analysis, which employed a tertiary care hospital database from Korea, indicated that ICI therapy is linked to a higher risk of developing incident diabetes than conventional chemotherapy, through a propensity matching analysis [70]. However, the study did not differentiate between T1DM and T2DM. In terms of ICI-T1DM, Hong et al. [71] reported four cases of ICI-T1DM, all of whom presented with ICI-induced DKA. Three cases were associated with the use of pembrolizumab, while one was caused by atezolizumab use [71]. Further studies with a sufficient number of participants should be conducted to estimate the risk of ICI-T1DM and identify predictive factors associated with this disease in Korea.
Current treatment guidelines on ICI-T1DM
The American Diabetes Association (ADA) guidelines for 2022 describe ICI-induced diabetes mellitus as a unique type of T1DM, an immune-mediated diabetes [72]. Immunotherapy, particularly ICIs, has resulted in unanticipated negative side effects, such as immune system activation, which led to the development of autoimmune diseases. T1DM can occur with DKA and low or undetectable C-peptide levels as markers of endogenous cellular activity [73,74]. Less than half of these patients have T1DM-associated autoantibodies, indicating a different pathobiology. This irAE occurs in less than 1% of patients receiving ICIs but is more common in those receiving medicines that block the PD-1/PD-L1 pathway, either alone or in combination with other ICIs [47]. Although the incidence of other high-risk HLA alleles is equivalent to that in the general population [47], 76% of patients are at a high-risk of the HLA-DR4 variant. To date, family history or autoantibodies cannot predict the risk; thus, all healthcare practitioners who administer these medications should be aware of this adverse effect and appropriately inform the patients. The American Association of Clinical Endocrinology mentions diabetes associated with ICIs as one of common forms of secondary diabetes; the guideline recommended that patients on ICI therapy should be monitored closely for early detection and therapy for hyperglycemia and the long-term development of diabetes [75]. A Consensus Report by the ADA and the European Association for the Study of Diabetes also briefly address that the development of profound insulin deficiency associated with the use of ICIs is an emerging issue in the management of T1DM in adults [76]. However, a universal consensus for this specific type of illness has not yet been reached. Additional epidemiological facts and evidence will provide a context for this distinct type of T1DM.
The treatment for ICI-T1DM is not different from that of classical T1DM; most patients require multiple insulin injections. However, the optimal glycemic target could be different from that of classical T1DM because ICI-T1DM patients have cancer and decreased life expectancy. Although there are no specific guidelines for ICI-T1DM, insulin therapy is necessary in ICI-T1DM patients, and clinicians should treat them as patients with T1DM and cancer, considering the risks and benefits of the treatment.
CONCLUSIONS
The use of ICIs that target PD-1/PD-L1 and CTLA-4 has improved cancer management; however, these medications can also cause autoimmune issues, such as ICI-T1DM, which occurs predominantly with PD-1 suppression. Patients with cancer who receive ICIs (anti-PD-1, anti-PD-L1, or anti-CTLA-4 antibodies) for the reduction of immune regulation and initiation of an immune response against tumor tissue are at risk of developing adverse effects, including acute T1DM, due to immune regulation loss combined with the activation of naive autoreactive T-cells. Thus, patients receiving ICIs, particularly PD-1/PD-L1 inhibitors, are advised to regularly monitor their glucose levels and check for early signs and symptoms of diabetes.
Notes
CONFLICTS OF INTEREST
Chang Hee Jung has been associate editor of the Diabetes & Metabolism Journal since 2022. He was not involved in the review process of this article. Otherwise, there was no conflict of interest.
FUNDING
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (grant numbers: NRF- 2020R1A2C1101977: Chang Hee Jung).
Acknowledgements
None