Obesity, Diabetes, and Increased Cancer Progression
Article information

Abstract
Rates of obesity and diabetes have increased significantly over the past decades and the prevalence is expected to continue to rise further in the coming years. Many observations suggest that obesity and diabetes are associated with an increased risk of developing several types of cancers, including liver, pancreatic, endometrial, colorectal, and post-menopausal breast cancer. The path towards developing obesity and diabetes is affected by multiple factors, including adipokines, inflammatory cytokines, growth hormones, insulin resistance, and hyperlipidemia. The metabolic abnormalities associated with changes in the levels of these factors in obesity and diabetes have the potential to significantly contribute to the development and progression of cancer through the regulation of distinct signaling pathways. Here, we highlight the cellular and molecular pathways that constitute the links between obesity, diabetes, cancer risk and mortality. This includes a description of the existing evidence supporting the obesity-driven morphological and functional alternations of cancer cells and adipocytes through complex interactions within the tumor microenvironment.
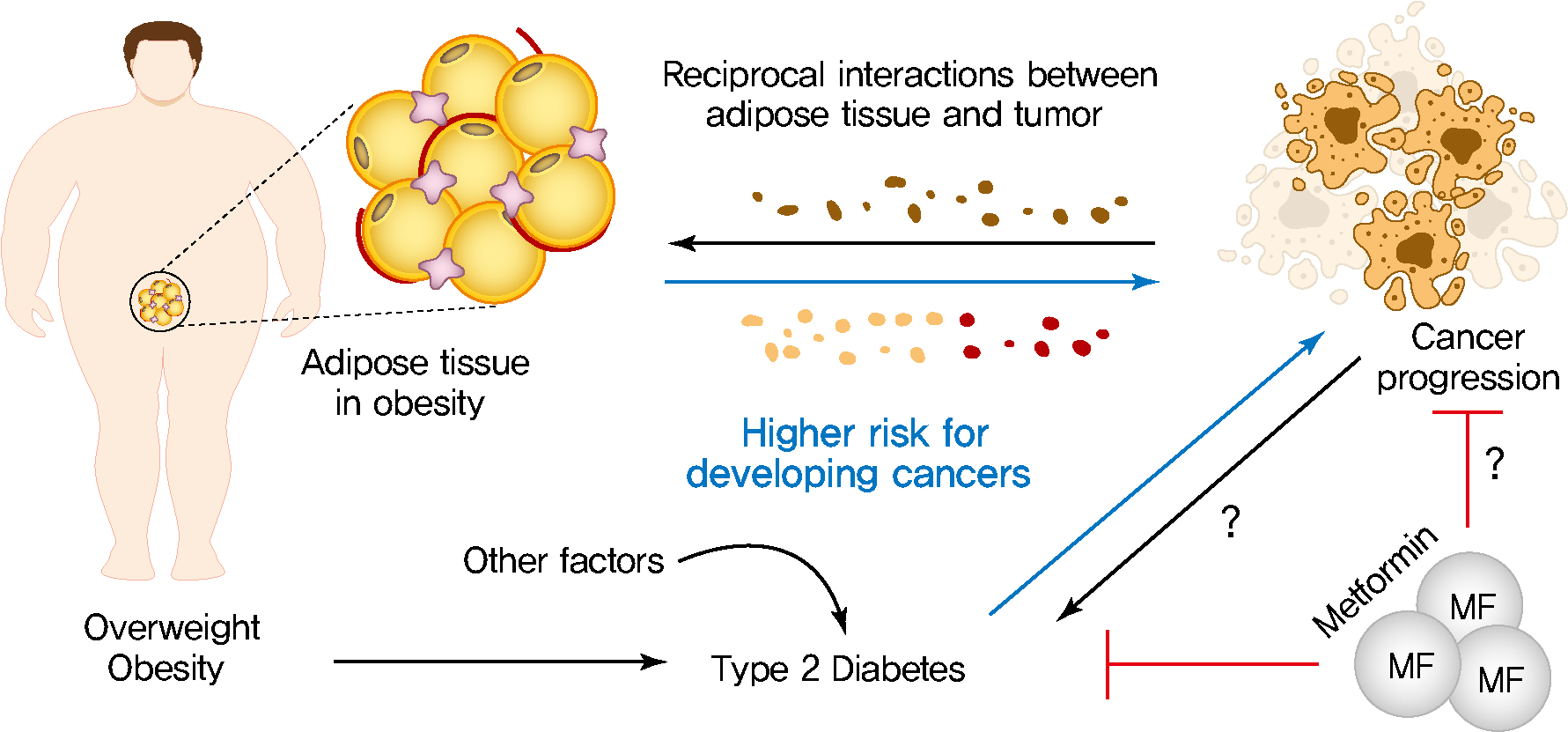
INTRODUCTION
The prevalence of obesity and diabetes has risen significantly for several decades and is expected to further increase gradually over the coming years [1]. Epidemiologically, individuals with obesity and diabetes are susceptible to an increased risk as well as a greater mortality rates for several types of cancers, such as endometrial, liver, pancreatic, colorectal, and breast cancer [2,3]. Therefore, understanding the molecular mechanisms underlying the epidemiological and mechanistic association between obesity, diabetes, and cancer has garnered significant attention as a therapeutic area of interest. Growing epidemiological evidence is hinting at a causal link between obesity, diabetes, and cancer [4-6]. Interestingly, it is the metabolic abnormalities associated with obesity and diabetes that may explain the link between metabolic dysregulation and cancer. Obesity leads to metabolic abnormalities of adipose tissue, affects the release of various hormones, adipokines, inflammatory cytokines, growth factors, enzymes, and free fatty acids [7,8]. These multiple metabolic substrates have been implicated as risk factors for cancer incidence and mortality [9-11]. In addition, the adipocyte-cancer cell crosstalk leads to morphological and functional changes in adipose tissue, resulting in changes to endocrine and paracrine signaling [10,12]. In turn, enhanced metabolic substrates released by altered adipose tissue physiology play a role in proliferation, invasion and metastasis of tumor cells [13]. Metabolic disturbances in type 2 diabetes mellitus (T2DM), such as hyperinsulinemia and dyslipidemia, have been proposed as causal links between diabetes and cancer [14,15]. High levels of insulin in hyperinsulinemia activate insulin/IGF-signaling and the subsequent activation of phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) signaling pathways promote cancer cell growth, survival, motility, and drug resistance [16-19]. In addition, hyperlipidemia leads to increased levels of cholesterol and non-esterified fatty acids (NEFAs), which are responsible for activation of oncogenic signaling pathways, membrane synthesis, and adenosine triphosphate (ATP) [14]. Here, we highlight the molecular mechanisms that point to a causal link between development of cancer and metabolic abnormalities that are associated with obesity and diabetes.
DYSFUNCTIONAL ADIPOSE TISSUE AND CANCER DEVELOPMENT
Obesity alters the metabolic profile of adipose tissue and leads to the increased secretion of numerous hormones, adipokines, inflammatory cytokines, growth factors, enzymes, and free fatty acids [7,8]. These adipose tissue-specific secreted factors contribute to the initiation and progression of several cancer types by driving metabolic reprograming of cells [20-22]. There is growing awareness of the essential role of adipocytesecreted factors from obese adipose for the development and progression of cancer.
Altered adipokine secretion
As a major endocrine organ, adipose tissue produces and secretes a variety of bioactive polypeptides, referred to as adipokines [23,24]. More than 600 adipocyte-enriched secretory factors have been discovered to date and adipokines play a critical role in maintaining glucose and energy homeostasis, as well as a range of metabolic pathways through communication with other organs [25]. The excess expansion of adipose tissue in obesity alters adipokine secretion and promotes chronic low-grade inflammation, thereby contributing to the development of metabolic disorders, including obesity and T2DM (Fig. 1) [26]. In addition, the dysregulation of adipose tissuespecific adipokines has an impact on the cellular physiology of various tumor cells and affects cancer cell growth, proliferation [27], migration, invasion [28], epithelial-mesenchymal transition (EMT) [29], angiogenesis [29], metastasis [29], and development of multidrug-resistance [9,21,30,31]. These altered adipokine profiles are associated with an altered metabolic state and directly provide substrates to help cancer cells meet their energy demands for the various biological processes mentioned above. Two of the most abundant and well-studied adipokines, adiponectin and leptin, have garnered significant attention in the field of cancer [32]. A number of studies have shown that adiponectin plays a protective role against obesityassociated cancer development and progression [33]. Adiponectin, a 30-kDa adipokine encoded by the ADIPOQ gene, is involved in regulating insulin sensitivity, glucose levels, as well as fatty acid breakdown [34,35]. Low serum adiponectin levels are associated with chronic inflammation and metabolic disorders, including T2DM, obesity, cardiovascular disease, and cancer development [35]. Several studies have shown that serum adiponectin levels are inversely associated with the risk of developing several types of cancer [36-38]. For example, low levels of adiponectin are associated with increased cancer risk, while the higher adiponectin levels are linked to a decreased cancer risk, and reduced cancer progression [39]. However, these effects may be very indirect with high adiponectin levels being a correlate to lower body weight, thereby decreasing the risk of tumor development. Other studies have shown that once an oncogenic mutation has occurred within a tissue, adiponectin can exert a positive effect on tumor growth [40]. Several mechanisms have been proposed to explain how adiponectin regulates cellular signaling pathways in cancer. Upon binding to its receptors, AdipoR1 and AdipoR2, and the possible engagement of the adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1), adiponectin regulates multiple downstream signaling pathways, including adenosine monophosphate-activated protein kinase (AMPK), mTOR, PI3K/protein kinase B (AKT), MAPK, peroxisome proliferator-activated receptor γ (PPARγ), signal transducer and activator of transcription 3 (STAT3), and nuclear factor-κB (NF-κB) [41-45]. Further studies are required to resolve the heterogeneous effect of adiponectin on tumor growth. Leptin is a 16-kDa multifunctional peptide hormone produced mainly by adipocytes and regulates food intake and energy expenditure to maintain body weight [46]. Plasma leptin concentrations increase in proportion to body fat mass, and elevated circulating leptin levels are a hallmark of obesity [32]. Growing evidence suggests that excess leptin and overexpression of its receptor (Ob-R) impacts the signaling pathways involved in cell proliferation, migration, invasion, metastasis, and EMT in breast cancer [47]. Leptin binds to Ob-R on breast cancer cells and within tumor tissue enhances several tumor cell responses via aberrant activation of multiple signaling pathways, such as the activation of the MAPK kinase (MEK)/extracellular signal-regulated kinase (ERK)1/2, Jak/STAT3, and PI3K/Akt signaling pathways [47]. Leptin and Ob-R are known to be expressed in estrogen receptor (ER)-positive breast cancer types and enhance ERα-dependent transcription to promote cell proliferation [48]. Interestingly, recent studies have begun to reveal that Ob-R is also highly expressed in hormone receptor-negative breast cancers, indicating that leptin and Ob-R may have a distinct role in the context of hormonereceptor negative breast cancers [49]. The functional interactions between leptin and Ob-R in the regulation of hormone receptor-negative breast cancers are highly intriguing and promise to be an excellent area of intervention in future studies. In addition, the generation and secretion of leptin also increased in epithelial tumor cells and tumor-associated stromal cells, suggesting that the production of leptin is not limited to adipocytes in the tumor microenvironment [50]. Importantly, multiple clinical studies have highlighted that elevated serum leptin levels are associated with cancer progression, metastasis, and poorer prognosis in patients with various other cancer types [47]. Therefore, given their key roles in various cancers, leptin and Ob-R are attractive therapeutic targets. As such, the inhibition of the leptin/Ob-R interaction and their clinical applications have attracted substantial interest and opened another avenue for treatment advances with the potential to dramatically impact breast cancer.
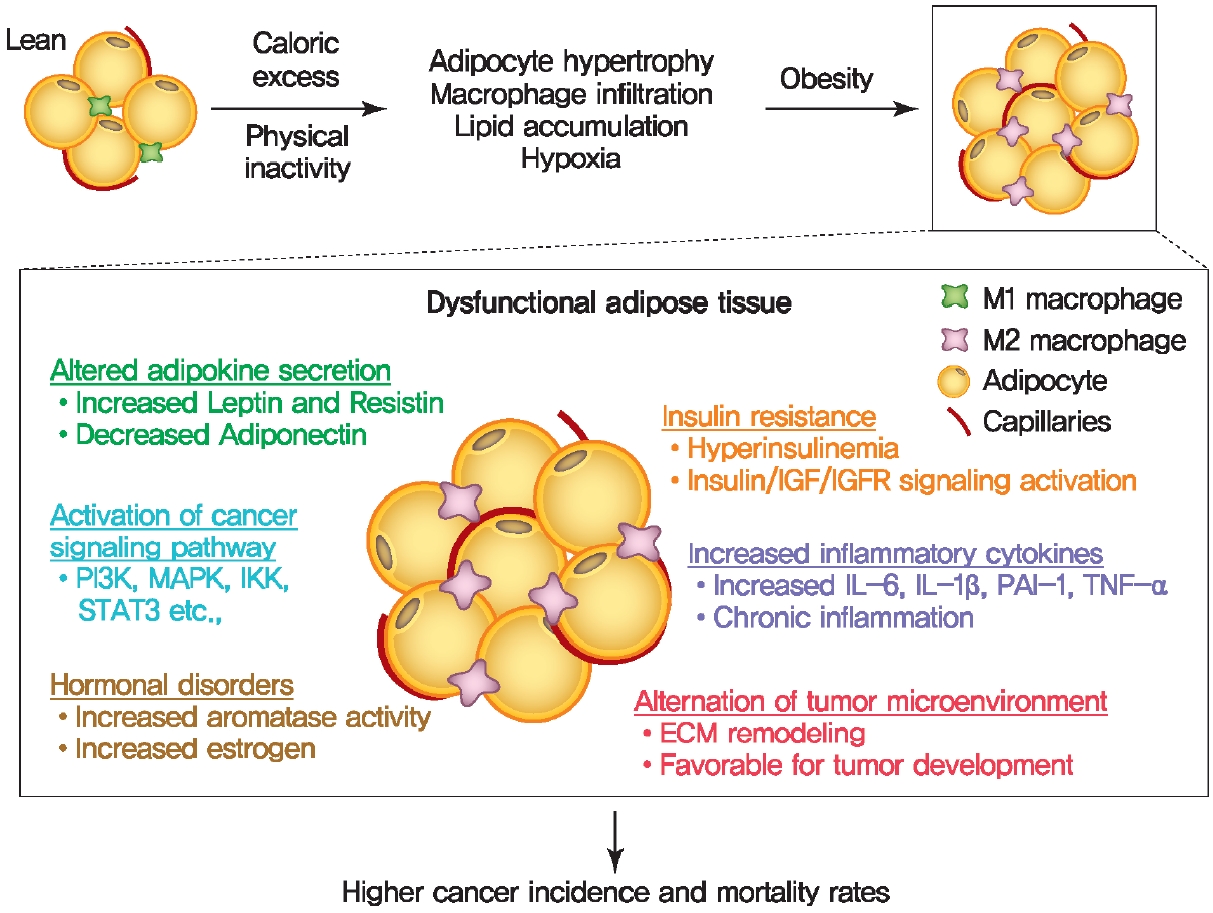
Proposed role of obesity-associated dysfunctional adipose tissue in tumor development and progression. Obesity-associated systemic metabolic disorders in adipose tissue contribute to the initiation and progression of cancer by producing endocrine and paracrine factors and alternation of tumor microenvironment. IGF, insulin-like growth factor; IGFR, insulin-like growth factor receptor; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; IKK, IkB kinase; STAT3, signal transducer and activator of transcription 3; IL, interleukin; PAI-1, plasminogen activator inhibitor 1; TNF-α, tumor necrosis factor-α; ECM, extracellular matrix.
Insulin resistance
Insulin is a hormone that is produced by β-cells in the pancreas and is primarily responsible for stimulating glucose uptake and storage to regulate glucose levels in blood [51]. Insulin also is involved in evolutionarily conserved pathways, such as cell growth, proliferation, and differentiation as well as protein and lipid synthesis, RNA and DNA synthesis. Mechanically, insulin binds to the insulin receptor (IR), a tyrosine kinase, that stimulates glucose uptake into metabolically active tissues such as skeletal muscle, adipose tissue and liver through enhanced translocation of the insulin-dependent glucose carrier 4 (glucose transporter 4 [GLUT4]) to the plasma membrane. Insulin resistance is clinically defined as the state in which a given concentration of insulin exerts a biological effect lower than expected. Impaired insulin-stimulated glucose uptake as well as reduced glucose oxidation and glycogen synthesis are known to be associated with insulin resistance [51]. Frequently, obesity is associated with development of insulin resistance (Fig. 1). Obesity may very well cause insulin resistance through promoting chronic inflammation in adipose tissue, and by increasing insulin secretion in the system, thereby activating multiple growth pathways [7]. However, insulin per se can also cause obesity due to its nature as a potently anabolic hormone. Chronic inflammation, excess insulin secretion and hyperactivation of growth pathways are closely associated with tumor development and progression [52]. Thus, obesity-induced insulin resistance plays a pathogenic role in contributing increased higher cancer incidence and cancer‐specific mortality.
Increased inflammatory cytokines
Obesity-associated chronic low-grade inflammation is known to be major factor in the development of metabolic disease and several types of cancer [9]. Chronic low-grade inflammation is characterized by increased infiltration and activation of immune cells along with increased local and systemic cytokine levels, leading to subsequent immune dysregulation in adipose tissue, liver, pancreas, skeletal muscle, and brain. Activation of several metabolic signaling pathways including IKKβ/NF-κB and c-Jun N-terminal kinase (JNK) pathways by excess nutrients leads to initiation of obesity-associated inflammation and a subsequent chronic low-grade inflammatory response (Fig. 1) [9]. In healthy and lean adipose tissue, M2 macrophages secrete antiinflammatory interleukin 10 (IL-10) cytokines, and M2 marker genes such as arginase 1 (Arg-1), fizzled, and Ym1 to maintain adipose tissue homeostasis [7]. In contrast to lean white adipose tissue, macrophages are recruited and infiltrated to white adipose tissue under obesogenic conditions and contribute to insulin resistance through promoting the secretion of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), IL-6, IL-1β, interferon γ (IFN-γ). Obese white adipose tissue can also shift the polarization of macrophages. During the course of obesity, the levels of the broadly defined pro-inflammatory M1 macrophages increase, leading to adipose tissue inflammation and insulin resistance by inhibiting IR signaling [7,53]. Enhanced secretion of macrophage-derived pro-inflammatory cytokines in obese adipose tissue is a major contributor to the pathogenesis of tumor development by activating several signaling pathways, such as the JAK/STAT pathway [9-11,54].
Hormonal disorders
Aromatase, belongs to cytochrome P450 family of monooxygenases, is an enzyme responsible for metabolism and cholesterol/steroid synthesis including estrogen biosynthesis [55]. Obesity has been associated with abnormally high aromatase expression in mammary tissues, which is synthesized in undifferentiated pre-adipocytes and adipose fibroblasts in postmenopausal women (Fig. 1) [2]. Obesity is associated with elevated expression and secretion of proinflammatory cytokines, such as TNF-α and IL-6 in adipose tissue, which contributes to increased aromatase expression in adipose tissue [56]. Elevated expression of aromatase in breast adipose tissue promotes the conversion of androgen to estrogen, leading to increased local estradiol concentrations and ERs activation [57-60]. Estrogens exert their biological functions via ERs that are ligand-dependent transcription factors that activate genes that are involved in cell proliferation, differentiation, apoptosis, and cell migration [61]. Dysregulated actions of ERα signaling are associated with breast cancer initiation and development [61,62]. ER+ tumors typically receive their growth signal from estrogen to promote cellular growth. In postmenopausal women, estrogen is mainly produced in peripheral tissues via the enzyme aromatase, thus inhibition of aromatase activity is often an effective treatment for ER+ tumors. Collectively, systemic and local estrogen production by aromatase in obese individuals contributes to the risk of hormone-dependent cancers. Therefore, inhibition of aromatase activity in the breast tissue is an established and effective treatment to curb the growth of estrogenreceptor-positive breast cancers.
Alternations in the tumor microenvironment
The mutual interactions between tumor cells and their adjacent microenvironment that result in a loss of tissue homeostasis imposed by changes in tissue architecture and polarity [63,64]. The tumor microenvironment has been recognized as a key contributor to many aspects of cancer development [65]. The extracellular matrix (ECM) and hypoxia have been widely demonstrated to play a critical role in the regulation of the tumor microenvironment. The main components of the ECM include various proteoglycans and fibrous proteins such as collagens, fibronectin, elastin, hyaluronan, and laminin. The ECM is one of the essential components of the tumor microenvironment, providing structural and mechanical support to tumor cells [66-68]. It is well established that tumor-driven ECM remodeling alters the biochemical and mechanical properties of the tumor microenvironment to create a favorable microenvironment for tumor cell growth, migration and metastatic progression [69-71]. The high oxygen demand of rapidly proliferating cancer cells causes an insufficient oxygen supply to the tumor, which becomes hypoxic [72]. Hypoxia is a well-known characteristic of the tumor microenvironment and promotes tumor angiogenesis, metastasis, reprogramming of metabolism, and resistance to therapy [73]. Emerging studies suggest that hypoxia also inhibits anti-tumor immune cells, which allows tumor cells to evade the immune system, thus establishing an immunosuppressive tumor microenvironment [74-77].
Obesity can influence the tumor microenvironment through dysfunctional adipose tissue and altered extracellular signals, thus promoting tumor growth, proliferation, angiogenesis, invasion, migration, and metastasis in breast cancer (Fig. 1) [13,78]. During obesity, vascular dysfunction is impaired and creates pockets of hypoxia. Adipose tissue hypoxia establishes a highly proinflammatory microenvironment, which is a favorable microenvironment for tumor promotion [65]. Dysfunctional adipose tissue leads to dysregulated adipokine production, preferentially releasing proinflammatory adipokines [23]. This adipokine imbalance is closely associated with insulin resistance, lipolysis and pro-inflammatory signaling pathways, contributing to a favorable microenvironment for tumor growth and progression. In addition, macrophage infiltration into obese adipose tissues promote ECM remodeling through elevation of several ECM components, thereby providing a tumor-friendly environment [21].
Activation of mitogenic signaling pathways
Obesity induces multiple intracellular signaling pathways through elevation of various signaling molecules including insulin, leptin and adiponectin (Fig. 1) [8]. Insulin and insulin-like growth factor-1 binding to their receptors (IR/IGF-1R), activating the PI3K/AKT/mTOR pathway, driving cell proliferation, invasion and metastasis [79-81]. Higher circulating levels of leptin secreted by obese adipose tissue binds to its cognate receptor (Ob-R), activating multiple signaling pathways, such as the JAK-STAT, PI3-kinase-AKT, and MAPK pathways, resulting in increased proliferation, migration and invasion of breast cancer cells [82,83]. In addition, bidirectional crosstalk between leptin and IGF-1 signaling plays an important role in obesity-associated breast cancer progression through epidermal growth factor receptor (EGFR) transactivation [84]. Adiponectin is known to inhibit tumor development and growth through activation of AMPK and subsequent downregulation of the MAPK pathway [36,85]. In addition, elevated glucose levels potentiate Wnt/β-catenin signaling through promoting the nuclear translocation of β-catenin [86]. Activation of the Wnt/β-catenin pathway enhances hypoxia-inducible factor 1α (HIF1α)-activated gene expression, thereby promoting cell survival and angiogenesis during hypoxia.
INTERACTION BETWEEN OBESITY AND CANCER
The crosstalk between adipose tissue and tumors in the obese state
Altered adipose tissue homeostasis in obesity can provide factors such as hormones, adipokines, and cytokines that assist cancer cells in acquiring the increased metabolic and energy demands mentioned above (Fig. 2) [7,9,56]. The crosstalk between cancer cells and adipocytes within the tumor microenvironment leads to further morphological and functional alteration of both cell types [10,78,87]. During interactions with cancer cells, adipocytes acquire phenotypic changes, including delipidation through multiple bioactive factors released by the cancer cells, resulting in a significantly decreased the expression of adipocyte-specific genes such as adiponectin, leptin, fatty acid-binding protein-4 (FABP4) [56]. Compared to normal adipocytes, this fibroblast-like phenotype of adipocytes is referred to as cancer-associated adipocytes (CAAs) [78]. Continuous stimulation of CAAs by paracrine signals from cancer cells leads to secretion of free fatty acids, inflammatory cytokines, adipokines, and growth factors, establishing a favorable tumor microenvironment which is an integral part of cancer development. Therefore, a greater understanding of functional crosstalk between CAA and cancer cell and new strategies for blocking this interaction may be an effective/attractive therapeutic target for treatment of cancer.
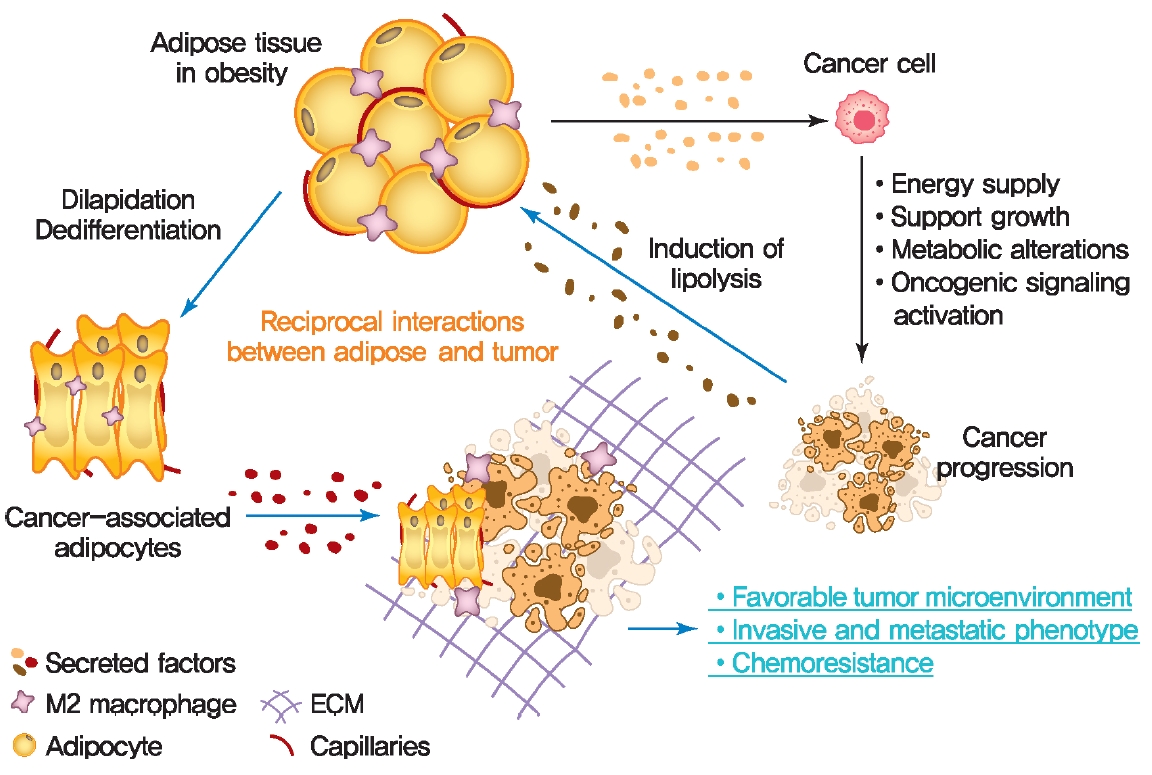
Adipocyte/cancer cell crosstalk in the obese state. Obesity increases the risk of developing of cancer by promoting multiple metabolic abnormalities of adipose tissue. In turn, cancer cells also induce morphological and functional changes in adipose tissue, which promote invasive and metastatic phenotypes of cancer. ECM, extracellular matrix.
Cancer metabolic reprogramming by adipokines
Cancer cells undergo reprogramming of glucose metabolism by increasing glucose uptake and glycolytic activity in the presence of oxygen (referred to as the Warburg effect) [88]. Growing evidence suggest that this glucose metabolic reprograming is driven by various adipose tissue-specific secreted factors. For example, leptin is known to promote glycolysis by stimulating key glycolytic enzymes and GLUTs, hexokinase, the M2 isoform of pyruvate kinase (PKM2), lactate dehydrogenase A (LDHA), and glucose-6-phosphate dehydrogenase (G6PDH) through PI3K/Akt activation [89-91]. Recent studies have revealed that p62 deficiency in adipose tissue supports nutrient availability for cancer cells through inhibition of energy-utilizing pathways in adipocytes [92]. Loss of p62 from adipose tissue leads to increased levels of osteopontin, an adipokine that subsequently enhances tumor cell fatty acid oxidation. Fatty acid oxidation provides the energy necessary for cancer cells to support tumorigenesis and cancer progression. These observations indicate a central role of adipose tissue-specific secreted factors in the regulation of cancer cell metabolic reprogramming (Fig. 2).
T2DM AND CANCER DEVELOPMENT
Diabetes mellitus (DM) is one of the most prevalent chronic metabolic disorder characterized by hyperglycemia [3]. Dysfunction of pancreatic insulin-producing β-cells and insulin resistance mainly leads to hyperglycemia, resulting in increased risk of T2DM development [93]. T2DM is most common form of DM, which accounts for around 90% of all cases of diabetes [93]. T2DM may be associated with increased risk, accelerated progression and greater mortality rates of several types of cancer such as liver, pancreatic, endometrial, colorectal, and breast cancer [3,94,95]. Anti-diabetic drugs, such as metformin, have recently attracted considerable interest and opened a promising avenue of research that has the potential for the treatment of breast and colorectal cancers due to their proposed anti-cancer properties [95-98]. In the following section, we discuss the various mechanism by which metabolic disturbances in T2DM, such as dyslipidemia, hyperinsulinemia, and hyperglycemia, may lead to the development of breast cancer.
Hyperinsulinemia
Hyperinsulinemia is characterized by chronically elevated insulin in the blood due to dysregulated insulin secretion and/or clearance [93]. Higher levels of the IR and increased circulating insulin are important factors that drive the risk of developing several cancers in T2DM patients (Fig. 3) [99-102]. Binding of insulin to IR leads to activating tyrosine kinase activity of the IR, resulting in tyrosine phosphorylation of IR substrate and subsequent activation of PI3K/Akt pathway which is responsible for most metabolic and mitogenic effects of insulin [103]. Activation of IR/PI3K/Akt signaling pathway directly phosphorylates and thus activates mTOR. mTOR then activates its downstream signaling pathway, which is involved in regulating cancer cell survival, proliferation, invasion, migration, differentiation, angiogenesis, and metastasis [103,104]. In addition, activation of IR/PI3K/Akt signaling pathway stimulates β-catenin translocation into the nucleus, increases the levels of vascular endothelial growth factor (VEGF), thus impacts the various tumor cell behaviors [105,106]. The IGFs are peptide hormones with high structural and functional similarity to insulin [103]. Binding of IGFs to their receptors (IGFRs) activates the intrinsic tyrosine kinase activity of receptor and initiates an intracellular signaling cascade, thereby play important roles in growth and development of many organs. Insulin and IGFs are able to bind to each other’s receptors, IR and IGFRs, and thus activate common downstream signaling including PI3K/Akt pathway and MAPK pathway [103,107]. High levels of insulin can lead to binding and activation of IGFRs signaling as well as an increase in the levels of bioactive IGFs, thus playing an essential role in cancer cell growth, survival, motility, and drug resistance.
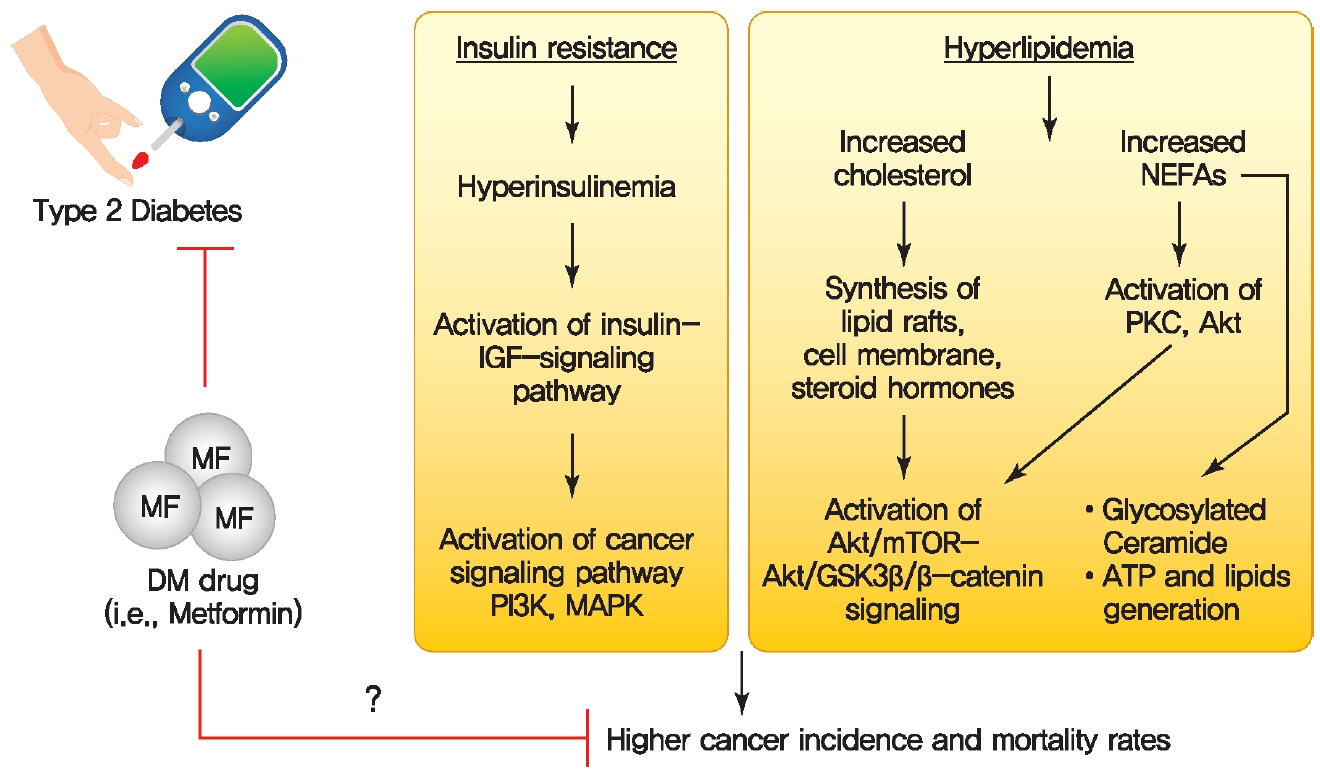
Possible molecular mechanisms for a direct link between diabetes and cancer. Diabetes-associated metabolic disturbances including hyperinsulinemia and dyslipidemia have been proposed as a causal link between diabetes and cancer. MF, metformin; DM, diabetes mellitus; IGF, insulin-like growth factor; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; NEFA, non-esterified fatty acid; PKC, protein kinase C; mTOR, mammalian target of rapamycin; GSK3β, glycogen synthase kinase 3β; ATP, adenosine triphosphate.
Hyperlipidemia
Dyslipidemia is often observed in patients with obesity, T2DM, and cardiovascular diseases [108]. Hyperlipidemia is characterized by alterations in circulating lipids with elevated low-density lipoprotein cholesterol, high levels of triglycerides, and low high-density lipoprotein cholesterol. Increased levels of cholesterol and NEFAs can indirectly act as signaling molecules involved in activation of intracellular signaling pathways such as the Akt/mTOR and Akt/glycogen synthase kinase 3β (GSK3β)/β-catenin oncogenic pathway (Fig. 3) [14]. 27-Hydroxycholesterol (27HC) is a primary metabolite of cholesterol, generated upon exposure to cytochrome P450 oxidase sterol 27-hydroxylase A1 (CYP27A1), an enzyme involved in regulating cellular cholesterol homeostasis [18]. 27HC acts as an ER agonist to activate ER related signaling mechanisms, such as the Akt/mTOR pathway or Akt/GSK3β/β-catenin, thereby stimulating cell proliferation and protein synthesis in ER-positive breast cancers (Fig. 3) [109,110]. High levels of CYP27A1 expression correlate with high-grade tumors in human breast cancer specimens, and inhibition of CYP27A1 reduces tumor growth in hormone-dependent breast cancers [111,112]. Interestingly, the expression of 27HC is different in various obesity models such as diet-induced obesity, db/db mice, and ob/ob mice. Therefore, future studies should address the how the dynamic changes of 27HC expression under different obesity conditions affect ER-positive breast cancer development [113]. The protein kinase C θ (PKCθ) is activated by elevated levels of serum NEFAs, which causes activation of Akt/mTOR and Akt/GSK3β/β-catenin oncogenic pathway [114,115]. Therefore, reducing the circulating levels of cholesterol and NEFAs and interfering activity of CYP27A1 and PKCθ present potential strategies for curbing breast cancer growth. Cholesterol is an integral component of membranes and lipid rafts in highly proliferative cancer cells [116]. In addition to the regulation of cellular signaling pathways, cholesterol also serves as the precursor for steroid hormone synthesis, which drives the initiation and progression of several cancers, indicating that cancer cells require cholesterol for tumor growth and survival [117]. Elevated NEFA levels in cancer are responsible for generation of ceramides. Ceramide glycosylation by glucosyl ceramide synthase (GCS) confers upon cancer cells multidrug resistance to cytotoxic anticancer agents [14,118]. In addition, NEFAs are also involved in ATP generation through the β oxidative pathway as well as synthesis of signaling and membrane lipids to support cancer cell proliferation and tumor growth [14].
The potential effect of diabetes medication metformin in cancer prevention
A series of studies have assessed whether the use of insulinsensitizing medications, such as metformin [95-98,119-124] and thiazolidinediones (TZDs) [119,120,125,126], may lower the incidence of cancer in diabetic patients (Fig. 3). Metformin is a first-line pharmacological treatment for T2DM and reduces hepatic glucose production by decreasing hepatic gluconeogenesis, resulting in increasing insulin sensitivity [127]. Multiple mechanistic studies with metformin in vitro and in animal models have elucidated the molecular mechanisms that underlie the action of metformin [127]. In particular, one major effect of metformin has been proposed to be the suppression of mTOR signaling pathway. Metformin reduces the circulating levels of insulin and IGF-1 in peripheral blood and activates liver kinase B1 (LKB1)/AMPK signaling pathways, leading to inhibition of the mTOR pathway, thus reducing cell proliferation, protein translation, and insulin levels [128]. However, the possible role of metformin as preventive agent against cancer in patients remains controversial and now is being extensively studied. A multiple population-based cohort study in patients with diabetes indicated that metformin therapy was not significantly associated with a reduced risk of cancer among patients with diabetes [129-131]. Further prospective mechanistic and large population-based cohort studies will be required to evaluate the effect of metformin therapy in cancer incidence.
Thiazolidinediones
TZDs, PPARγ agonists, are insulin-sensitizing medications that reduce insulin resistance and decrease hepatic glucose production agents in T2DM [132]. In addition to their essential metabolic actions in T2DM, TZDs have been shown to exert anti-tumor effects by affecting the cell cycle, apoptosis and cell differentiation in several types of cancer including breast and colon cancer [133]. However, multiple clinical studies revealed that treatment with TZDs was not associated with a significantly anti-neoplastic effect in several cancers [134-136]. Therefore, further large-scale prospective studies will likely need to resolve these inconsistencies.
CONCLUSIONS
The growing prevalence of obesity and diabetes are closely linked to an increased incidence and mortality of many types of cancer [2-4,9,56,94]. The multiple metabolic factors and disorders linking obesity and T2DM with cancer that are broadly discussed in this review may be important for determining the therapeutic potential of cancer treatment. Altered adipose tissue metabolism in obesity results in altered levels of multiple factors, such as hormones, adipokines, inflammatory cytokines, growth factors, enzymes, and free fatty acids to help cancer cells satisfy their metabolic and energy demands [7,13,24]. Adipocytes also acquire phenotypic changes through multiple bioactive factors released by surrounding cancer cells, resulting in significantly increased secretion of many metabolic substrates [30,78,87]. This adipocyte/cancer cell crosstalk within the tumor microenvironment leads to further morphological and functional alterations of both cell types, which is gradually being recognized as an integral part of cancer development and progression. However, many important questions remain regarding the molecular fingerprint and the biological roles of such a crosstalk in promoting cancer development and progression. Therefore, (1) a more in-depth understanding of how adipocytes interact with tumor cells and contribute to cancer development and progression is required; (2) identification of specific targets that can serve as promising avenues to limit tumor proliferation need to be identified; (3) determination of new strategies to block this interaction could be an effective/attractive therapeutic strategy in the treatment of cancers. T2DM is the most common form of diabetes that appears to be associated with an increased risk of several types of cancer due to its associations with multiple metabolic disturbances [94,95]. Various metabolic disturbances involved in the development of diabetes, such as dyslipidemia and hyperinsulinemia, are well established to lead to the development of a cancer-conducive microenvironment. Based on these observations, a series of studies suggest that the use of anti-diabetic drugs, e.g., metformin, may have the potential to reduce the cancer incidence and/or mortality in diabetic patients. Mechanistically, metformin reduces the circulating levels of insulin and IGF-1, thus inhibiting the mTOR pathway and subsequent cancer cell proliferation [128]. Given the potential of metformin for cancer therapy, metformin has attracted increased attention and opened new avenues in cancer treatment due to their proposed anti-cancer properties. In contrast, a recent a population-based cohort study indicated that metformin use was not associated with significantly decreased risk of cancer among patients with diabetes [129-131]. Based on large population-based cohort studies, further insights into the potential utility of metformin will be required to evaluate the effect of metformin therapy in cancer incidence.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
Authors were supported by US National Institutes of Health grants R01-DK55758, R01-DK- 127274, R01-DK099110, RC2-DK118620 and P01-AG051459 to Philipp E. Scherer.
Acknowledgements
We thank Dr. Christine Kusminski for comments and extensive editing of the manuscript.