New Perspectives on Diabetic Vascular Complications: The Loss of Endogenous Protective Factors Induced by Hyperglycemia
Article information
Abstract
Diabetic vascular complications are among the leading causes of morbidity and mortality in diabetic patients. In the past, many studies have focused on the mechanisms of hyperglycemia-induced chronic vascular complications via the formation of toxic metabolites such as oxidative stress, advanced glycosylated end products, persistent activation of protein kinase C, and increased sorbitol concentrations. However, vascular complications result from imbalances caused by increases in systemic toxic metabolites, such as those that occur under conditions of hyperglycemia and dyslipidemia, and by reductions in endogenous protective factors such as insulin, vascular endothelial growth factor, and platelet derived growth factor. This review outlines some of the evidence supporting the importance of enhancing endogenous regenerative factors.
The vascular complications of diabetes mellitus affect many organ systems, including the retina, kidney, nerve, and cardiovascular system. These serious complications are the leading causes of mortality and morbidity in diabetic patients.
Diabetic vascular complications result from imbalances caused by increases in the toxic effects of systemic metabolic abnormalities such as hyperglycemia, dyslipidemia, and hypertension, and reductions in the regenerative effects of endogenous protective factors such as insulin, vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), nitric oxide (NO), and antioxidant enzymes (Fig. 1). In the past, many studies on the mechanisms of diabetic complications have focused on the mechanisms by which hyperglycemia might lead to the chronic vascular complications via the formation of toxic metabolites such as oxidants and advanced glycosylated products. These mechanisms include increases in oxidative stress, persistent activation of protein kinase C (PKC) and other signaling pathways, increased sorbitol concentrations through the aldose reductase pathway, the elevated formation of advanced glycosylation end products, and increased flux through the hexosamine pathway [1]. However, few studies have evaluated the importance of endogenous protective factors or the inhibitory effects of hyperglycemia in neutralizing these protective factors during the initiation and progression of diabetic complications. This review outlines some of the evidence supporting the importance of preventing and delaying the progression of diabetic complications. Further, the lack of success in finding effective clinical therapeutics to neutralize the toxic effects of hyperglycemia could be due to the need for enhancing protective factors.
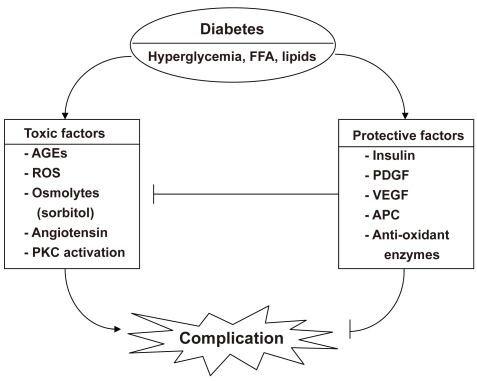
Diabetes induces an imbalance between toxic and protective factors to cause complications. FFA, free fatty acid; AGE, advanced glycosylated end product; ROS, reactive oxygen species; PKC, protein kinase C; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor; APC, activated protein C.
Hyperglycemia-induced cellular apoptosis is a common pathology of many diabetic complications such as for retinal pericytes, renal podocytes, and vascular endothelial cells. In the case of diabetic retinopathy, accelerated pericyte apoptosis is one of the earliest and most specific findings of diabetic complications. Enge et al. [2] reported that PDGF-B or PDGF receptor-β knockout mice exhibited pericyte apoptosis and retinal microvascular abnormalities similar to the early stages of diabetic retinopathy, indicating that PDGF-B is a very important survival factor for retinal pericytes. However, the level of PDGF-B expression was observed to be elevated in diabetic state compared with that in non-diabetic animal [3]. Thus, it was not clear whether pericyte loss results from PDGF-B abundance or PDGF-B resistance. Recently, Geraldes et al. [4] clearly demonstrated that hyperglycemia induced a persistent activation of PKC-δ, which leads to PDGF resistance in the retina. Under normal glucose condition, PDGF-B stimulated DNA synthesis, inhibited cellular apoptosis, and increased p-AKT and p-ERK activation in retinal pericytes. However, PDGF-induced activation of p-AKT and p-ERK signaling was blunted by hyperglycemic levels, which was restored in cells expressing dominant negative PKC-δ or in PKC-δ knockout mice. Geraldes et al. also identified SHP-1, a tyrosine kinase, as a cellular target of PKC-δ and p38α that inhibited the survival action of tyrosine kinase growth factor receptors such as PDGFR-β, by an NF-KB-independent pathway. Therefore, hyperglycemia induces activation of PKC-δ - p38α MAPK - SHP-1, which leads to the inhibition of PDGF-related survival actions that cause pericyte apoptosis in diabetic retinopathy. This study has identified SHP-1 as a new potential therapeutic target, to be inhibited as a treatment for diabetic vascular complications.
Another survival factor is VEGF, which is one of the most important endogenous angiogenic polypeptides that respond to hypoxia under normal physiological conditions. The expression of VEGF is increased in the retina, causing diabetic proliferative retinopathy; however, its abnormally low expression in the myocardium and peripheral lesions causes poor collateral vessel formation in peripheral tissues. During angiogenesis, VEGF interacts with several other angiogenic factors, playing an important role in cell proliferation, differentiation, migration, cell survival, NO production, release of other growth factors, and sympathetic innervation. VEGF may have actions in non-endothelial cells. VEGF is also produced by renal podocytes. Podocytes and fenestrated endothelial cells are two major cells of the glomerular filtration barrier. Mesangial cells are located between the capillary loops and provide structural support. The apoptosis of podocytes is an early event of diabetic nephropathy and a likely cause of proteinuria. Sison et al. [5] reported that VEGF-A released by podocytes was required for glomerular endothelial cell migration, differentiation, and survival through cell-specific gene targeting. The tyrosine kinase receptors for VEGF (VEGFR-1 and VEGFR-2) were expressed by glomerular endothelial cells. Their podocyte-specific, heterozygous, conditional knock out of VEGF-A mice showed a loss of endothelial cell fenestrations, endothelial cell necrosis, loss of podocyte foot process, and a marked loss of mesangial cells. This suggests that the paracrine effects of VEGF are crucial for glomerulus development and maintaining the function of the glomerular filtration barrier.
Microalbuminuria is an early detectable clinical abnormality in diabetic glomerulopathy. Metabolic pathways activated by hyperglycemia, glycated proteins, hemodynamic factors, and oxidative stress are key players in the pathogenesis of diabetic nephropathy. A variety of growth factors and cytokines are then induced through complex signal transduction pathways. Podocyte-derived VEGF is an angiogenic factor whose expression is increased in animal models of diabetic kidney disease [6]. Controversies exist on the effect of the diabetes-induced enhanced expression of VEGF in the renal glomeruli. Some have suggested that VEGFs act in a novel autocrine signaling mode to induce the podocytopathy of diabetes, especially the genesis of albuminuria. Thus, treatment with anti-VEGF antibodies may attenuate glomerular basement membrane thickening and progression of diabetic nephropathy. However, recently Eremina et al. [7] reported proteinuria, hypertension, and renal failure in several patients treated with anti-VEGF agent, bevacizumab. Pathologic findings from kidney biopsies were prominent endothelial swelling, red cell fragments, mesangiolysis, and thrombi in capillary loops which were similar to those in podocyte-specific VEGF-A knockout mice. The study also suggested that anti-VEGF therapy inhibits the VEGF action of podocytes and results in endothelial damage and thrombotic microangiopathy.
An alternative explanation is that the increased VEGF expression in the kidney is a compensatory action to protect the glomeruli from the toxic effects of hyperglycemia. This means that the increase of VEGF is a compensatory response to overcome ischemic or other stressful injury which can induce VEGF resistance in the renal glomeruli by diabetes.
In contrast with diabetic microvascular complications, VEGF expression is reduced in diabetic heart and macrovasculature. Ventricular tissues from muscle insulin receptor knockout mice, which lack insulin receptors in the myocardium, have significant reductions in insulin but not IGF-I signaling, VEGF expression, and vascular density before and after ischemia versus controls [8]. This suggests that impaired insulin signaling has decreased VEGF expression and aggravated ischemic injury. Thus, VEGF is an important angiogenic and survival cytokine in protecting micro- and macro-vascular tissues from conditions such as hyperglycemia.
Insulin is another important survival factor for vascular endothelial cells. Insulin has been shown to have both anti-atherosclerotic and pro-atherosclerotic actions in vascular cells. The anti-atherosclerotic actions of insulin include decreased endothelial cell apoptosis and reactive oxygen species, and increased NO by activation of endothelial NO synthase and heme oxygenase-1. In contrast, insulin has also been reported to have pro-atherosclerotic actions such as increasing endothelin-1 and plasminogen activator inhibitor-1, and enhancing the migration and growth of vascular smooth muscle cells. In diabetes, hyperglycemia can selectively inhibit insulin's anti-atherosclerotic actions, while hyperinsulinemia, as observed in insulin-resistant type 2 diabetes mellitus, can enhance insulin's pro-atherosclerotic effects to accelerate the prevalence of atherosclerotic diseases [9]. Rask-Madsen et al. [10] recently reported that loss of insulin signaling in endothelial cells accelerated atherosclerosis in apolipoprotein E-null mice. Double knockout mice with a deletion of the endothelial insulin receptor and apolipoprotein E gene (EIRAKO) were created using the Cre/loxP system to achieve tissue-specific deletion of these genes. Despite retaining systemic insulin sensitivity, glucose tolerance, plasma lipids, and blood pressure were not different between the EIRAKO and control mice; however, more severe atherosclerotic lesions, impaired vasodilatation, and increased VCAM-1 expression were observed in the EIRAKO mice vs. the apolipoprotein E-null mice. Therefore, improving insulin sensitivity in the endothelium may decrease the risk for atherosclerosis in insulin resistance and diabetes states.
Clinical evidences are also available in support of the importance of endogenous protective factors. Recently, we reported that a significant number of type 1 diabetes mellitus patients with a duration of diabetes of 50 years or longer do not have significant retinopathy and nephropathy (Joslin Medalist Study) [11]. This study demonstrated a lack of association between degree of hyperglycemia and prevalence of microvascular complications, although as a group, glycemic control in the study participants is very good, with a mean HbA1c of 7.3%. These results suggest that unknown endogenous protective factors are greatly reducing microvascular complications in this group of type 1 diabetic subjects of extremely long duration. If the protective factors could be found from the study of these patients, we may be able to prevent or delay microvascular complications in all diabetic patients.
In summary, new therapeutic approaches for diabetic vascular complications need to focus on decreasing the toxic factors of diabetes, but also need to focus on increasing the action of protective factors.