O-GlcNAc Modification: Friend or Foe in Diabetic Cardiovascular Disease
Article information
Abstract
O-Linked β-N-acetyl glucosaminylation (O-GlcNAcylation) is a dynamic post-translational modification that occurs on serine and threonine residues of cytosolic and nuclear proteins in all cell types, including those involved in the cardiovascular system. O-GlcNAcylation is thought to act in a manner analogous to protein phosphorylation. O-GlcNAcylation rapidly cycles on/off proteins in a time scale similar to that for phosphorylation/dephosphorylation of proteins. Several studies indicate that O-GlcNAc might induce nuclear localization of some transcription factors and may affect their DNA binding activities. However, at the cellular level, it has been shown that O-GlcNAc levels increase in response to stress and augmentation of this response suppresses cell survival. Increased levels of O-GlcNAc have been implicated as a pathogenic contributor to glucose toxicity and insulin resistance, which are major hallmarks of type 2 diabetes and diabetes-related cardiovascular complications. Thus, O-GlcNAc and its metabolic functions are not yet well-understood; focusing on the role of O-GlcNAc in the cardiovascular system is a viable target for biomedical investigation. In this review, we summarize our current understanding of the role of O-GlcNAc on the regulation of cell function and survival in the cardiovascular system.
INTRODUCTION
The addition of O-linked β-N-acetyl glucosamine (O-GlcNAc) to the hydroxyl groups of serine and threonine residues on target proteins is a dynamic regulatory post-translational modification of nuclear and cytosolic proteins [1]. It is involved in a wide range of biological processes such as nuclear transportation, transcription and translation, signal transduction, cytoskeletal reorganization, proteasomal degradation, and apoptosis [2,3]. In terms of abundance and protein distribution, O-GlcNAc is often considered to be analogous to the phosphorylation of proteins [3]. Just as protein phosphorylation is regulated by hundreds of kinases and phosphatases, O-GlcNAcylation is also tightly regulated by an enzymatic process. So far, only O-GlcNAc transferase (OGT) has been shown to catalyze the O-GlcNAcylation of proteins and one enzyme, N-acetyl-glucosaminidase (O-GlcNAcase or OGA), catalyzes its removal [4,5]. The end-product of the hexosamine biosynthesis pathway (HBP), uridine diphosphate-N-acetyl glucosamine (UDP-GlcNAc), acts as a substrate for the O-GlcNAcylation of proteins [6]. Approximately 2-5% of the total intracellular glucose enters the HBP and is ultimately converted to UDP-GlcNAc. The metabolism of glucose via the HBP is essential for the synthesis of O-GlcNAc, which serves as a metabolic sensor that attenuates the cellular response to extracellular stimuli based on the energy state of the cell [7]. Several studies have demonstrated that an acute increase in O-GlcNAc synthesis improves cell survival, while a sustained increase in O-GlcNAc levels has been implicated as a pathogenic contributor to glucose toxicity and insulin resistance, major hallmarks of type 2 diabetes (T2D) and diabetes-related cardiovascular complications [8].
In this review, we summarize our understanding of the role of O-GlcNAc in mediating both the adverse effects of diabetes as well as cellular protective mechanisms in the cardiovascular system.
CHARACTERIZATION OF O-GLCNACYLATION OF PROTEINS
Under normal conditions, two to five percent of the glucose taken up by cells is consumed by the HBP, and entrance of glucose into the HBP is regulated by glutamine: fructose-6-phosphate amidotransferase (GFAT), which converts fructose-6-phosphate to glucosamine-6-phosphate using glutamine as an amine donor. Glucosamine-6-phosphate is then metabolized via various intermediates, leading to the synthesis of UDP-GlcNAc [9]. In addition, UDP-GlcNAc can be also increased by addition of exogenous glucosamine, which enters into cells via the glucose transporter system and is phosphorylated to glucosamine-6-phosphate by hexokinase, instead of the GFAT-dependent HBP, leading to an increase in UDP-GlcNAc levels [10]. Using UDP-GlcNAc as a substrate, OGT catalyzes the transfer of GlcNAc via an O-linkage to specific serine and threonine residues on target proteins [11]. OGT activity/O-glycosylation is vital for life, confirmed by the observation that deletion of the OGT gene was embryonic lethal [12]. The level of O-GlcNAcylation of nucleocytosolic proteins is also regulated by O-GlcNAcase, which catalyzes the removal of the sugar moiety from the target proteins. Similar to the processes of phosphorylation/dephosphorylation, O-GlcNAcylation on the target proteins rapidly activates/deactivates the activities of proteins involved in many aspects of cellular processing. At the same time, O-GlcNAcylated proteins can also be modified via phosphorylation, which modulates the functions of proteins by influencing protein-protein interactions and protein localization (Fig. 1) [13].
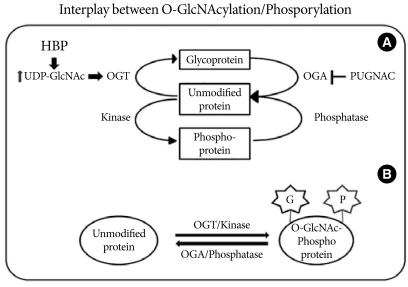
Interplay between O-GlcNAcylation and protein phosphorylation. (A) O-GlcNAc modification is strongly dependent on the concentration of UDP-GlcNAc produced by the hexosamine biosynthetic pathway (HBP). OGT uses UDP-GlcNAc as a substrate for GlcNAcylation on protein serine and threonine residues. O-GlcNAcase (OGA) removes the O-GlcNAc moiety from O-GlcNAc-modified proteins. PUGNAC inhibits the activity of O-GlcNAcase. (B) O-GlcNAc modification is analogous to protein phosphorylation/dephosphorylation. Interplay between GlcNAcylation and phosphorylation can affect the activities or stabilities of the proteins, and both processes can occur on the same protein at proximal sites.
O-GLCNACYLATION, DIABETES, AND INSULIN RESISTANCE
Insulin resistance plays a major pathophysiological role in T2D and is tightly associated with major health problems, including obesity, heart diseases, and dyslipidemia [14]. Marshall et al. [15] demonstrated that flux through the HBP is responsible for the development of insulin resistance and remarked that the development of insulin resistance requires glucose, insulin, and glutamine. The role of glutamine in the development of insulin resistance has been found to occur via the regulation of GFAT, the rate limiting enzyme in the HBP [16]. Inhibition of GFAT with either azaserine or 6-diazo-5-oxo-norleucine abrogated the effects of hyperglycemia on the development of insulin resistance [17]. Studies have shown that an increase in cellular UDP-GlcNAc and O-GlcNAcylation levels on target proteins due to high glucose and glucosamine treatments leads to oxidative stress and endoplasmic reticulum stress, which have been shown to cause chronic inflammation and insulin resistance [18]. Blocking of the removal of O-GlcNAc using the OGA inhibitor, (2-acetamido-2-deoxy-D-glucopyranosylidene) amino-N-phenyl carbamate (PUGNAC) results in a decrease in insulin-response glucose uptake in 3T3-L1 adipocytes [19]. In insulin target cells, there appears to be a negative feedback regulation of glucose transport via the flux of glucose through the HBP [20]. Insulin resistance and the corresponding decrease in glucose uptake are correlated with a defect in the translocation of the glucose transporter GLUT4 to the plasma membrane [20]. Recent studies point to phosphatidylinositol-4, 5-bisphosphate (PIP2)-assisted remodeling of filamentous actin at the inner leaflet of the plasma membrane (cortical F-actin) as another crucial step in insulin-stimulated GLUT4 translocation [21]. Bhonagiri et al. [22] showed that exposing 3T3-L1 adipocytes to excess HBP flux leads to a reduction in PIP2 content in the plasma membrane, with a concomitant loss in cortical F-actin, which affects insulin-induced GLUT4 translocation into the plasma membrane. In addition to the effects on GLUT4 translocation, it has been reported that there are associations among HBP, insulin resistance, and impairment of Akt/PKB signaling [23]. Yang et al. [24] demonstrated that upon insulin stimulation, OGT is rapidly recruited from the nucleus to the plasma membrane by PIP3 through a region adjacent to the catalytic domain II at the carboxyl terminus of the OGT and demonstrated a potential mechanism by which Akt is presumably able to be O-GlcNAcylated by the PIP3-recruited OGT. Based on these findings, impairment of O-GlcNAcylation regulation is reflected by a decrease in glucose uptake through insulin resistance.
O-GLCNACYLATION INDUCES VASCULAR DYSFUNCTION
Hyperglycemia and insulin resistance are major causative factors for T2D and its vascular complications. High circulating glucose concentrations alter functions of the vascular endothelial cells and the underlying smooth muscle cells [25]. Hyperglycemia can induce vascular complications via several different mechanisms, and one of the mechanisms is an increase of the HBP flux of glucose and O-GlcNAcylation of target protein [8]. Hyperglycemia-induced oxidative stress inhibits glyceraldehydes 3-phosphate dehydrogenase, resulting in a decrease in glycolytic flux and an increased glucose flux in the HBP [26]. This increased flux of glucose may influence vascular remodeling as it can modulate the cell proliferation or cell death of different vascular cell types, such as pericytes, endothelial cells, or smooth muscle cells.
O-GLCNACYLATION ON VASCULAR SMOOTH MUSCLE CELLS
Vascular smooth muscle cell (VSMC) dysfunction is a major risk factor of diabetic cardiovascular disease. VSMCs are highly specialized cells whose principal functions are the contraction of blood vessels and the regulation of blood vessel tone-diameter, which regulate the blood pressure and the blood flow, respectively. Under diabetic conditions, an increased flux of glucose through the HBP has been proposed to cause vascular disease. It has been observed that prolonged exposure to high glucose leads to the increase of GFAT expression in VSMCs [27], indicating that GFAT is possibly involved in the development of the diabetic vascular complications. Inhibition of GFAT activity using 6-diazo-5-oxonorleucine decreases the hyperglycemia-induced tumor growth factor-alpha (TGF-α) expression in VSMCs, suggesting that the adverse effects of hyperglycemia in VSMCs are mediated by the HBP. Hall et al. [28] demonstrated that expressions of GLUT1 and GLUT4 are increased in the neointima of the aorta after balloon injury. Increased proliferation and decreased apoptosis of VSMCs provides a possible linkage with the increased risks of restenosis and atherosclerosis in patients with diabetes. Akimoto et al. [29] found that the pattern of O-GlcNAc modification of proteins changed when rat aortic smooth muscle cells (RASMCs) were cultured in medium containing a high concentration of glucose. High glucose also elevates both the expression and activity of OGT [29]. In addition, high glucose and glucosamine also induced an increase in the expression of growth factors in RASMCs [30]. The effects of O-GlcNAc on cell growth and division may also contribute to the increase in VSMC proliferation seen in diabetes [31]. Treatment of VSMCs with high glucose increases the expressions of TGF-α and basic fibroblast growth factor, leading to abnormal proliferation of smooth muscle cells, a characteristic associated with atherosclerotic lesions [30]. It has been reported that an elevated glucose concentration in VSMCs causes an increase in the activity of protein kinase C (PKC), known to modulate the expression of several growth factors including TGF-α [32]. McClain et al. [30] also found that treatment of cells with phorbol ester to repress PKC and treatment of staurosporine to inhibit PKC did not affect the ability of glucose or glucosamine to activate the TGF-α promoter. Hattori et al. [33] found that hyperglycemia-induced activation of PKC was involved in the activation of TNFα-induced NF-κB, a transcription factor involved in the pathogenesis of atherosclerosis. Recently, Yang et al. [34] demonstrated that Thr352 of the P65 subunit of NF-κB was O-GlcNAcylated, which results in increases in its transcriptional activity and half-life within the nuclei of VSMCs cultured under high glucose conditions. They also found that attenuation of O-GlcNAcylation by over-expression of OGA inhibits the high glucose-induced NF-κB activation in VSMCs. On the other hand, over-expression of OGT along with OGA inhibitor increases NF-κB transcriptional activity. These results suggest that O-GlcNAc modification of transcription factors presumably modulates their activities.
As described above, excess glucose flux through the HBP results in elevated UDP-GlcNAc levels in VSMCs, which may have the capacity to modify and regulate vascular reactivity. Proteins with an important role in vascular function, such as endothelial nitric oxide synthase (eNOS), sarcoplasmic reticulum Ca2+-ATPase, phospholipase C, PKC, and phosphoinositide-3-kinase, are also targets for O-GlcNAcylation, suggesting that O-GlcNAcylation may play a critical role in vascular reactivity [35]. In isolated rat thoracic aortic rings, increasing O-GlcNAc formation via PUGNAC blunted vascular relaxation by acetylcholine and augmented vasoconstriction in response to phenyleprine [36]. In addition, treatment with ST060266, an OGT inhibitor, inhibits the U46619-induced blood vessel contraction which depends on Rho kinase (RhoK) activity, suggesting that RhoK is also modified by GlcNAcylation (Kim DH and Jeoung NH, unpublished data). The contractile response of vascular smooth muscle is mediated through myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP) activations, which are initiated by the Ca2+-calmodulin interaction that causes phosphorylation of the myosin light chain (MLC) [37]. Clark et al. [38] demonstrated impaired calcium cycling via transcriptional inhibition of sarcoplasmic Ca2+-ATPase in OGT-over-expressing cardiomyocytes. It was demonstrated that membrane depolarization promotes OGT activation, leading to induction of O-GlcNAcylation of total proteins, which can be reduced by inhibition of either voltage-gated Ca2+ channels or Ca2+-calmodulin-dependent kinase IV (CAMIV), suggesting that OGT activity is regulated by Ca2+ influx and CAMIV-dependent phosphorylation. Interestingly, CAMIV has been shown to phosphorylate and activate OGT both in vivo and in vitro [39]. Smooth muscle cells also contain Ca2+-independent mechanisms to regulate contractibility. Removal of the phosphate group by MLCP results in inactivation of MLC and promotion of smooth muscle relaxation. MLCP consists of two subunits; one catalytic subunit of type 1 protein phosphatase (PP1cδ) and a non-catalytic subunit [40]. Recently Cheung et al. [41] found that MYPT1, a known targeting regulatory subunit of PP1cδ, is O-GlcNAcylated and also acts as a substrate for OGT. The substrate specificity of OGT is also regulated by MYPT1, which dephosphorylates OGT under different conditions. Based on the above findings, it is clear that O-GlcNAcylation on specific vascular proteins has an important role in the regulation vascular reactivity, and further research is necessary to determine the impact of O-GlcNAcylation on vascular reactivity.
O-GLCNACYLATION ON ENDOTHELIAL DYSFUNCTION
Endothelial cell dysfunction has emerged as a key component in the pathophysiology of cardiovascular abnormalities associated with diabetes [42]. Normal endothelial production of nitric oxide by eNOS plays a pivotal role in the regulation of vascular tone and remodeling, the inhibition of platelet aggregation and adhesion to the vascular wall, as well as the synthesis and secretion of extracellular matrix proteins, and proliferation of VSMCs [43]. Endothelial cells are sensitive to hyperglycemia because of their poor ability to regulate intracellular glucose [44]. In cultured bovine aortic endothelial cells, hyperglycemia caused an increase in mitochondrial superoxide production in association with an increase in O-GlcNAc on eNOS and a reciprocal decrease in phosphorylation at Ser1177, the site responsible for activation of the enzyme [45]. Inhibition of GFAT by antisense oligonucleotides blocks the O-GlcNAcylation of eNOS, confirming the role of increased HBP flux of glucose. This O-GlcNAc-induced decrease in eNOS activity was associated with increases in matrix metalloproteinase (MMP) activity and expression combined with decreased tissue inhibition of metalloproteinase (TIMP) expression [46]. An imbalance between MMPs and TIMPs has been implicated in atherosclerosis-related complications in diabetes [46]. Du et al. [47] demonstrated that hyperglycemia-induced expression of plasminogen activator inhibitor-1 (PAI-1) in endothelial cells was associated with an increase in O-GlcNAc levels on Sp1, which increases expression of TGF-1β, a potent inducer of extracellular matrix protein synthesis, leading to proliferation of VSMCs and fibroblasts. High glucose also induces O-GlcNAc modification of Sp3 and up-regulates angiopoietin-2 gene expression in the microvascular system [48]. This leads to the induction of ICAM-1 and VCAM-1 expressions and sensitization of microvascular endothelial cells to the proinflammatory effects of TNFα. Increased O-GlcNAcylation of Sp1 and Sp3 also regulates the endothelial cell-specific expression of vascular endothelium growth factor receptor [49]. These observations suggest that chronically elevated O-GlcNAc level represents a common mechanism underlying the adverse effects of hyperglycemia. The vascular effects of HBP/O-GlcNAc signaling are just beginning to be understood, and a more direct link between increased O-GlcNAc levels and vascular dysfunction remains to be discovered (Fig. 2).
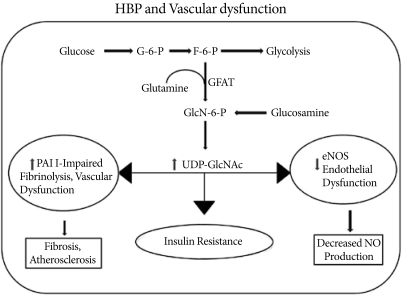
A fraction (2-5%) of the glucose entering a cell is directed into the hexosamine biosynthesis pathway (HBP) pathway. GFAT (glutamine: fructose-6-phosphate amidotransferase) uses glutamine to convert fructose-6-phosphate into glucosamine-6-phosphate, which is then used for the synthesis of UDP-GlcNAc in the cell. Activation of the HBP pathway acts through Sp1 sites to increase plasminogen activator inhibitor-1 (PAI-1) and tumor growth factor-alpha expressions. At the same time, O-GlcNAc modification of endothelial nitric oxide synthase (eNOS) decreases its activity and nitric oxide (NO) production.
O-GLCNACYLATION AND CARDIOVASCULAR PROTECTION
Many studies have shown that a loss of control of the dynamics of O-GlcNAcylation interferes with the normal progress of fundamental events and may be implicated in the appearance of pathological states. However, since OGT is essential for cell viability and is highly conserved from an evolutionary perspective, its presence has to convey some survival advantage to cells and organisms [12]. It has been supported by a number of studies demonstrating that an acute increase in O-GlcNAc synthesis improves tolerance to a variety of stressful stimuli and increases cell survival [50,51]. When the O-GlcNAc response was inhibited by decreased OGT expression, cell viability also decreased, whereas augmentation of O-GlcNAc levels with PUGNAC increased cell survival [52]. Glucose uptake in cells has been linked to the capacity of cells to respond and survive deleterious cellular conditions. Even though the sustained hyperglycemia in T2D is clearly associated with the adverse effects at the cellular level, the acute increases in circulating glucose level and of glucose utilization in tissues have beneficial effects on cell survival. It has been reported that increased glucose utilization protects against a wide range of injuries like trauma, sepsis, shock, cardiac surgery, and myocardial infarction [50]. During acute heat stress conditions, an increase in O-GlcNAc levels enhances cell survival, whereas inhibition of GFAT decreases O-GlcNAc levels in the cells, leading to an increase in cell death [51]. Increasing the O-GlcNAc level results in cells that are more thermo-tolerant, which results in an increase in the expressions of heat shock protein 40 (HSP40) and HSP70 induced by the O-GlcNAc signaling pathway [53].
Acute activation of O-GlcNAc formation inhibits acute inflammatory and neointimal responses to endoluminal vascular injury in vivo [54]. Further, increases in O-GlcNAc-modified proteins decrease the expressions of inflammatory mediators and the infiltration of leukocytes at the injured endoluminal carotid artery. Since augmentation of O-GlcNAc levels attenuates the activation of inflammatory mediators in acute cardiovascular injury, it is possible that an increased O-GlcNAc level in diabetes may be beneficial for its ability to attenuate the proinflammatory response. Glucosamine treatment during resuscitation improved cardiac function by reducing the trauma-hemorrhage-induced ICAM-1 expression, NF-κB expression, and NF-κB DNA binding activity in the heart [55], suggesting that acutely increased O-GlcNAc level may reduce the NF-κB-mediated inflammation.
In isolated neonatal cardiomyocytes, hypoxia-reperfusion causes a transient increase in O-GlcNAc level and improves cell viability. Treatment of neonatal cardiomyocytes with glucosamine, hyperglycemia, or PUGNAC significantly increases O-GlcNAc level, improves cell viability, and decreases necrosis and apoptosis [56]. Schaffer et al. [57] reported that high glucose significantly reduces hypoxia-induced apoptosis and necrosis in isolated cardiomyocytes via decreased Ca2+ overload. Interestingly, cardiac mitochondria isolated from PUGNAC-treated mice and also OGT-over-expressing neonatal rat cardiomyocytes are resistant to Ca2+ induced mitochondrial membrane transition pore (mPTP) formation, a critical step in the initiation of apoptosis and cell death [58]. Recently, Champattanachai et al. [59] found that glucosamine, OGT over-expression, and O-GlcNAcase inhibition protect neonatal cardiomyocytes from ischemia-reperfusion injury via an increased level of mitochondrial bcl-2, a key regulatory protein for cell survival. A glucosamine-induced increase in bcl-2 inhibits mPTP opening via direct interaction with voltage-dependent anion channel 1α, one of the putative components of mPTP (Fig. 3).
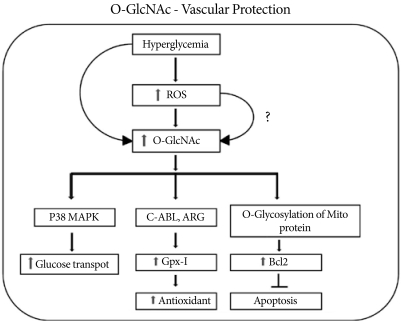
Hyperglycemia-induced activation of O-linked N-acetyl glucosamine activates GPX1 and its binding to c-Abl and Arg kinases and protects the cell via the antioxidant response. Along with an increase in the antioxidant response, increased O-GlcNAcylation activates p38 MAPK phosphorylation with increased glucose transport. Increased O-GlcNAc formation also increases the level of mitochondrial Bcl2, which inhibits apoptosis and protects the cell during stress.
Thus, acute activation of metabolic pathways leading to an increase in O-GlcNAc level is an endogenous stress-activated response, and augmentation of this response improves short term protection in the cardiovascular system. Induction of prosurvival factors such as HSP or attenuation of the inflammatory response improves long term cardiovascular protection. Further studies on the function of O-GlcNAc-modified proteins in complex signaling networks will provide answers surrounding cell survival in response to stress.
CONCLUSIONS
In this review, we summarized the data supporting the beneficial effects related to acute increases in O-GlcNAc level, as well as the adverse effects produced by chronic activation. This suggests that the initial response to an acute stress induces protective processes and improves survival; as the load increases, the continued activation of the pathway results in the development of pathophysiology. Thus, altering the O-GlcNAc levels in vascular tissues may represent a novel therapeutic approach for the treatment of diabetic cardiovascular disease.