Mitochondrial Toxins and Healthy Lifestyle Meet at the Crossroad of Hormesis
Article information

Abstract
Mitochondrial function is crucial for the maintenance of cellular homeostasis under physiological and stress conditions. Thus, chronic exposure to environmental chemicals that affect mitochondrial function can have harmful effects on humans. We argue that the concept of hormesis should be revisited to explain the non-linear responses to mitochondrial toxins at a low-dose range and develop practical methods to protect humans from the negative effects of mitochondrial toxins. Of the most concern to humans are lipophilic chemical mixtures and heavy metals, owing to their physical properties. Even though these chemicals tend to demonstrate no safe level in humans, a non-linear dose-response has been also observed. Stress response activation, i.e., hormesis, can explain this non-linearity. Recently, hormesis has reemerged as a unifying concept because diverse stressors can induce similar stress responses. Besides potentially harmful environmental chemicals, healthy lifestyle interventions such as exercise, calorie restriction (especially glucose), cognitive stimulation, and phytochemical intake also activate stress responses. This conceptual link can lead to the development of practical methods that counterbalance the harm of mitochondrial toxins. Unlike chemical hormesis with its safety issues, the activation of stress responses via lifestyle modification can be safely used to combat the negative effects of mitochondrial toxins.
INTRODUCTION
Chronic exposure to low-dose chemical mixtures has emerged as a risk factor for many aging-related chronic diseases because these chemicals can disturb the metabolic, hormonal, nervous, and immune systems at doses to which humans are environmentally exposed [12345]. Although endocrine-disrupting chemicals (EDCs) are the main class of chemicals that have gained attention in this regard [12], new terms covering broader harmful pathways have recently been coined, such as metabolism-disrupting chemicals [3], mitochondrial function-disrupting chemicals [4], and homeostasis-disrupting chemicals [5].
A significant number of molecular mechanisms link individual compounds to biological effects in in vitro or in vivo studies [1234]. However, as humans are exposed to complex chemical mixtures, it is difficult to determine the specific compounds linked to specific health conditions in humans [5]. Although a large number of epidemiological studies linking specific compounds with specific outcomes continue to be published, there is always the possibility that the compound measured in each study is a surrogate marker for many compounds that tend to coexist in the environment and humans. Therefore, human studies focusing on environmental exposure should always consider chemical mixtures.
Chemical mixtures affecting mitochondrial function require special attention because proper mitochondrial function is crucial for the maintenance of cellular homeostasis. As mitochondrial dysfunction can result in the progressive development of numerous pathological conditions, including type 2 diabetes mellitus (T2DM), dementia, and cancer, as well as aging [6], it is reasonable to assume that chronic exposure to chemical mixtures affecting mitochondrial function can increase the risk of a wide range of chronic diseases.
The following two types of mitochondrial toxicity are possible: (1) direct mitochondrial damage due to high dose exposure to individual compounds and (2) subtle but chronic mitochondrial damage due to low-dose chemical mixture exposure. Although there are many early toxicological studies on the former exposure condition, in this article, we will focus on the latter, because it is more relevant to the current situation in humans.
In this hypothetical review, we argue that hormesis, a long debated concept in the field of chemical toxicology [7], should be revisited to (1) explain the non-linear dose-response relationships of mitochondrial toxins observed within a background environmental exposure range and (2) develop practical strategies that can protect humans from the negative effects of mitochondrial toxins. As extensive reviews on chemical-induced mitochondrial toxicity (including molecular mechanisms) have recently been published in a special issue of toxicology [8], those topics are not the focus of this review.
MITOCHONDRIA STRESS RESPONSE
As a highly dynamic organelle, the primary function of mitochondria is to generate ATP during oxidative phosphorylation (OXPHOS) to meet cellular energy requirements, and also to activate a wide range of biochemical pathways including the biosynthesis of macromolecules such as heme and steroid hormones [9]. Recent advances in mitochondrial research have revealed that the mitochondria also play a critical role as signaling organelles [9].
Under both physiological and stressor conditions, the mitochondria constantly communicate with the nucleus and cytoplasm, in response to cellular needs and their own dysfunction [10]. Multiple mechanisms such as respiratory chain dysfunction, mitochondrial protein unfolding, mitochondrial membrane potential changes, and mitochondrial fission/fusion are involved in this communication [10]. They eventually promote a wide range of adaptive defense mechanisms including increased mitochondrial biogenesis and antioxidant defenses, augmented cytoprotective responses, xenobiotics detoxification, and enhanced metabolism, which can contribute to the extension of a healthy life [1112].
The most critical molecules involved in the communication between the mitochondria and other parts of the cell are mild reactive oxygen species (ROS) such as the superoxide anion and hydrogen peroxide [13]. Within healthy cells, the primary production source of signaling mild ROS is mitochondrial OXPHOS [14]. Therefore, the continuous production and scavenging of mild ROS during OXPHOS is crucial to maintain homeostasis under physiological condition [13], creating oscillatory dynamics in mitochondrial biological reactions [15].
Under stress conditions, ROS production increases. However, organisms can use this signal to activate adaptive defense mechanisms [1112]. In this situation, the challenge is how to quickly and safely scavenge increased ROS, not the increased ROS itself. If there is chronic impairment in the scavenging process, adaptational and compensatory stress responses can gradually be turned into pathological conditions.
The essential role of mild ROS as signaling molecules means that a very low production of mild ROS can be as harmful as high ROS production (Fig. 1C). This situation differs from the traditional linearity concept of ROS-induced harm based on the free radical theory of aging [16] (Fig. 1A); it also differs from a biphasic curve that only contrasts the benefits of mild ROS versus the harm of high ROS (Fig. 1B) [17].
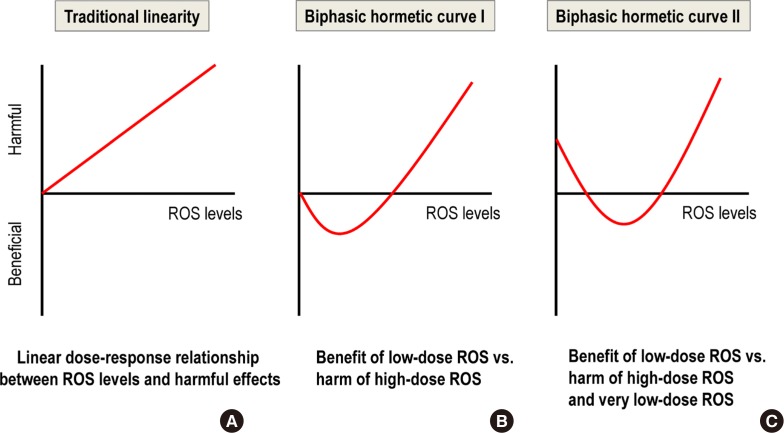
Comparison of three reactive oxygen species (ROS) dose-response curves. (A) Traditional linearity. (B) Biphasic hormetic curve I. (C) Biphasic hormetic curve II.
PERSISTENT CHEMICAL MIXTURES AS MITOCHONDRIAL TOXINS
Some pharmaceutical drugs have long been considered mitochondrial toxins [18]. Emerging evidence indicates that common environmental chemicals can also cause mitochondrial dysfunction. A wide range of environmental chemicals are suspected to be mitochondrial toxins acting via multiple mechanisms, including dynamic changes in mitochondrial fusion and fission, structure/membrane damage, and respiratory chain dysfunction [192021].
However, mitochondrial dysfunction owing to chemical exposure does not always lead to pathological conditions because the mitochondria can induce multiple stress responses as adaptational mechanisms, in particular when stressors are transient [10]. In fact, transient mitochondrial stress due to mitochondrial toxin exposure can even be beneficial via the augmentation of adaptive defense mechanisms, as discussed in the “mitochondria stress response” section [11]. However, this benefit can diminish as mitochondrial stress becomes chronic.
Therefore, in humans, the most concerning chemicals may be mitochondrial toxins with long half-lives [22]. Examples include lipophilic chemical mixtures such as persistent organic pollutants (POPs) and heavy metals. In particular, POPs and heavy metals are known to accumulate within the mitochondria due to their lipophilic and cationic properties [19]. In the development of mitochondria-targeting drugs, the most effective mitochondria drug delivery method is to covalently link a lipophilic cation, such as an alkyl triphenylphosphonium moiety, that can achieve more than 100- to 1,000-fold higher mitochondrial concentrations than that of the medium [23].
Other examples of chronic exposure to mitochondrial toxins are air pollution and cigarette smoking, both of which involve exposure to complex mixtures of gaseous and particulate components that vary in chemical composition [24]. In fact, POP-like lipophilic compounds and heavy metals are widely detected as the primary components of particulate matter (PM) [2526].
As POPs and heavy metals are mainly metabolized through glutathione (GSH) conjugation [2728], chronic exposure to very low-doses of these chemicals can lead to the depletion of cellular GSH reserves [2930]. As GSH constitutes the primary antioxidant mitochondrial defense [31], chronic GSH depletion due to chronic exposure to these chemicals can render the mitochondria vulnerable, even without directly affecting mitochondrial function. Therefore, chronic GSH depletion may be one of the mechanisms involved in the harmful effects caused by mitochondrial toxins, even at a very low-dose in humans. However, GSH depletion can be reversed as the dose is closer to the hormetic zone as we discuss in the following section, because increased GSH synthesis is one of the typical features of hormetic responses [32].
HORMESIS CAN EXPLAIN THE NON-LINEAR RESPONSE IN THE LOW-DOSE RANGE
In a range of environmental exposures, a non-linear response with no safe level has been widely observed for common mitochondrial toxins with different outcomes in human studies [33]. Examples include lead and intelligence quotient (IQ) deficits [34], lead and cardiovascular disease mortality [35], PM2.5 and T2DM [35], PM2.5 and cardiovascular disease mortality [36], benzene and leukemia [37], and POPs and T2DM [38]. The risk of disease increases at doses that are considerably below those recommended as the upper limits by regulatory agencies; however, the risk increase slows down or flattens with increasing doses [5]. This feature sharply contrasts with the linear dose-response relationship observed at high doses.
This non-linear response observed in epidemiological studies has long been dismissed as statistical artifacts or biases by researchers [39]. However, substantial evidence supports the biological plausibility of a non-linear response. Although endocrine disruption has been the most studied mechanism for the non-monotonic dose-response relationships of individual compounds [40], it is difficult to explain the non-linear response observed in epidemiological studies due to the unpredictable net effect of the real-world EDC mixture [5]. The more plausible explanation is the activation of stress responses at certain chemical levels (hormesis) [41]. In fact, chemical hormesis is conceptually identical with the stress responses discussed in the “mitochondria stress response” section. Environmental chemicals are just one of the stressors that can induce stress responses.
In the field of toxicology, the traditional concept of hormesis only contrasts a “beneficial low-dose zone” versus a “toxic high dose zone” [7]. This construction fails to recognize possible harmful effects at the sub-hormetic low-dose level [42]. This sub-hormetic zone can explain the negative effects observed in the range close to zero exposure observed in several epidemiological studies. However, as the dose approaches the hormetic zone, it can result in no further risk increase with increasing dose.
Despite the extensive evidence of hormesis in in vitro and in vivo models [43], whether hormetic chemical effects can be observed in humans has long been debated. However, hormetic effects of environmental chemicals would not manifest as a clear benefit in humans subjected to chronic exposure to chemical mixtures. No further risk increase associated with increasing dose and the activation of counteracting forces observed in the real world can be considered as indirect evidence of hormesis.
Namely, within the low-dose range, no-safe level with a non-linear response, which has been observed with many common environmental chemicals, may be the result of a combination of reactions in the “sub-hormetic harmful zone” and “hormetic beneficial zone” (Fig. 2, zone A+B). The range is in contrast with linearity, which is commonly observed in the high-dose toxicity range (Fig. 2, zone C).
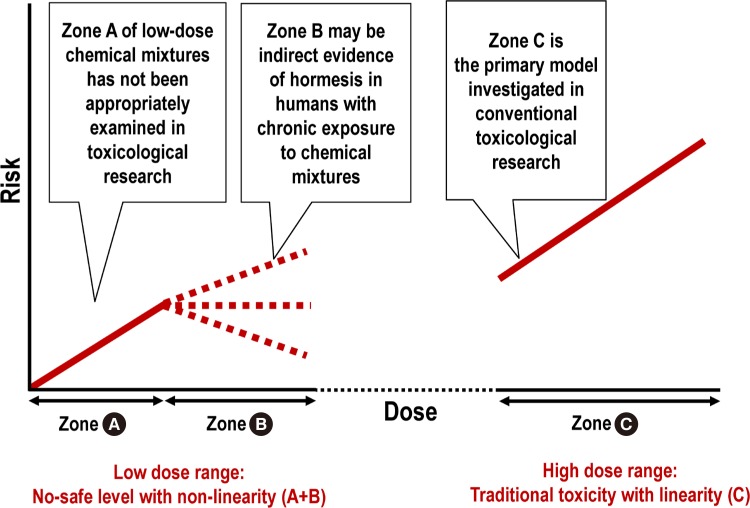
Comparison between the low-dose range (zone A+B) and high dose range (zone C). Chronic exposure to low-dose chemical mixtures can be harmful (zone A), but the risk can flatten or decrease (zone B) through the activation of stress responses (i.e., hormesis). The traditional biphasic hormesis curve contrasts only a beneficial low-dose (zone B) with a harmful toxicity dose (zone C) but does not properly examine the negative effects in the sub-hormetic zone (zone A).
CURRENT APPROACHES CANNOT EFFECTIVELY PROTECT HUMANS FROM THE HARM OF LOW-DOSE ENVIRONMENTAL CHEMICALS
As information regarding the low-dose effects of environmental chemicals accumulates, identifying effective methods to protect humans from low-dose range exposure is crucial. Although policies or interventions to reduce exposure to common pollutants to a minimum or to zero range can be considered because there seems to be no-safe level, these approaches cannot be solutions for the low-dose range for the following reasons.
First, it would be practically impossible to reduce the levels of common pollutants to near zero in the environment. For example, lead exposure reduction after the introduction of a policy banning lead gasoline is cited as a successful attempt to regulate environmental pollutants [44]. However, while the reduction from 10 to 5 mg/mL is possible with the development of effective policy, the reduction from 5 to 1 mg/mL is another story. Exposure sources become increasingly diverse and vague as exposure levels decrease; therefore, further reduction through regulation becomes harder in the low-dose range.
Second, even if the reduction of one or two environmental pollutants to practically zero was possible, our environment would still be unsafe because they are only a tiny part of the whole picture. Humans are currently exposed to a tremendous number of low-dose environmental chemicals. They include indispensable exposure sources such as air, water, and food. The chemical exposure problem is exacerbated by the fact that contemporary human adipose tissue already stores lipophilic chemical mixtures that are continuously released into the circulation by controlled and uncontrolled lipolysis [45].
Therefore, alternative methods should be explored to protect humans from the negative effects of low-dose exposure [46]. Conventional approaches to mitigate the damage of environmental chemicals, such as strict regulation and the avoidance of exposure sources, can work against the toxicity of several individual chemicals at high doses, but the negative effects of low-dose chemical mixtures require additional approaches to protect humans [46].
MITOCHONDRIAL TOXINS AND HEALTHY LIFESTYLES CAN MEET AT THE CROSSROAD OF HORMESIS
Importantly, hormesis has now reemerged as a unifying concept. With understanding of the role mitochondria play in stress responses to maintain homeostasis (discussed in the “mitochondria stress response” section), a wide range of stressors can induce similar stress responses that result in beneficial outcomes [1112]. These stressors range from environmental chemicals or radiation, which were initially considered to be harmful, to healthy behaviors, which were initially considered to be beneficial [47].
Stress responses can be a reasonable underlying mechanism to explain the non-linearity of the dose-response curve in the low-dose range of mitochondrial toxins such as POPs, heavy metals, and air pollution. This concept defines hormesis from the viewpoint of environmental chemicals. However, well-accepted healthy behaviors, such as exercise, calorie restriction (especially glucose), cognitive stimulation, and phytochemical intake, can also activate similar stress responses although all of them do not work through the mitochondria [174849]. In contrast to hormesis related to exposure to synthetic chemicals or radiation, they are called as mitohormesis [50] or xenohormesis [49]. Detailed molecular mechanisms of these behaviors in relation to stress responses can be found elsewhere [515253].
Based on this conceptual link of stress responses, herein, we suggest practical ways to decrease the negative effects caused by chronic mitochondrial toxin exposure as a hypothesis. In the case of transient exposure to mitochondrial toxins, unless they are highly toxic, they can act as stressors to activate hormesis and thus living organisms may not reveal any pathological phenotype (Fig. 3A). However, in the case of chronic exposure to mitochondrial toxins, they tend to demonstrate their negative effects at very low-doses with non-linearity as epidemiological studies have reported. In this situation, the activation of stress responses with the exposure to other stressors can be useful to mitigate that negative effects of chronic mitochondrial dysfunction (Fig. 3B).
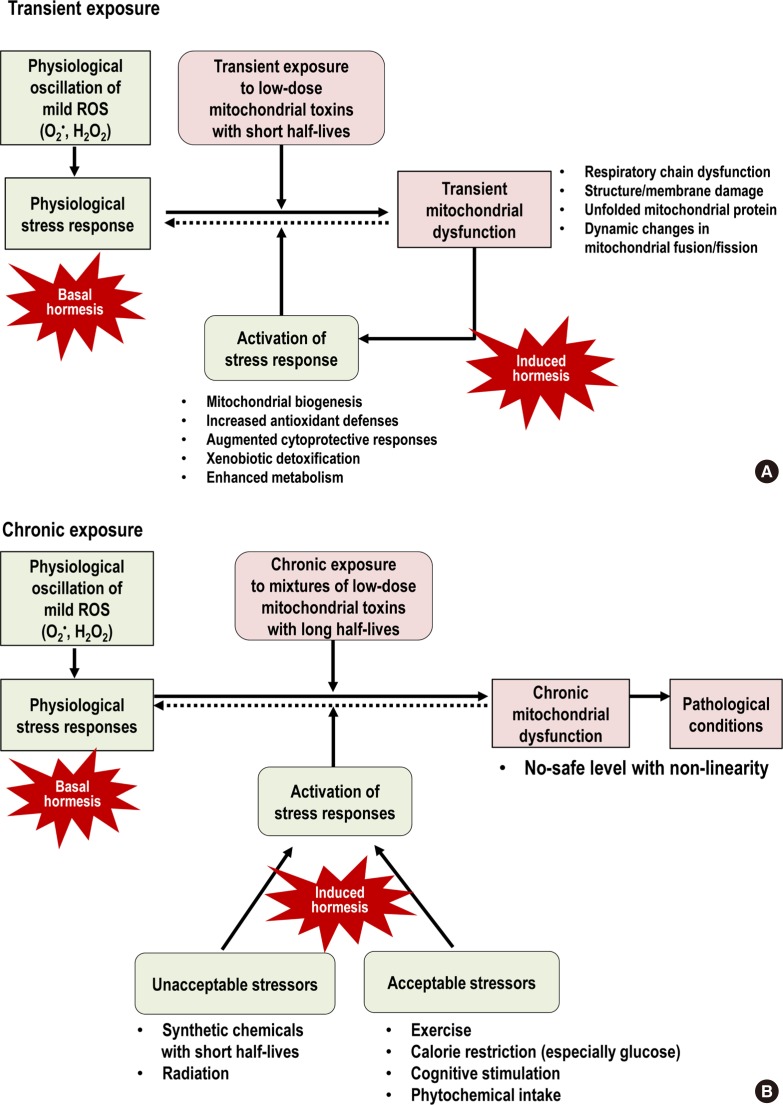
Comparison between (A) transient exposure and (B) chronic exposure to low-dose mitochondrial toxins. Transient exposure can be overcome via the activation of stress responses in which mitochondrial toxins themselves play a role as stressors. However, chronic exposure can lead to the development of pathological conditions through chronic glutathione (GSH) depletion and other mitochondrial dysfunction. In this situation, the activation of stress responses using other stressors is needed to mitigate negative effects of chronic mitochondrial dysfunction. Among various stressors, exercise, calorie restriction (especially glucose), cognitive stimulation, and phytochemicals are examples of stressors that are considered acceptable to the public. ROS, reactive oxygen species.
Among many stressors that can activate stress responses, from the viewpoint of human application, they can be classified into the following two groups: stressors unacceptable to public versus stressors acceptable to public. For example, it would be difficult for humans to deliberately use the hormetic effects of environmental chemicals or radiation which were initially considered to be harmful to public. There are safety related concerns and uncertainty on the doses that demonstrate hormetic effects in humans. However, the induction of stress responses through mitohormesis or xenohormesis-activating lifestyles can be safely used because these behaviors are generally well-accepted by public as healthy. Thus, hormesis-activating lifestyle can be helpful in decreasing the possible harm of low-dose exposure to environmental chemicals.
Meanwhile, there is another crucial aspect of hormesis to consider because optimal health requires physiological stress responses which are maintained by continuous production and scavenging of mild ROS during OXPHOS in the mitochondria. We call it “basal hormesis,” contrasting with “induced hormesis” that is activated by external stressors (Fig. 3). As we discussed in the “persistent chemical mixtures as mitochondrial toxins” section, chronic exposure to mitochondrial toxins can induce the impairment of basal hormesis through chronic GSH depletion (Fig. 3B).
There is direct experimental evidence suggesting that the activation of stress responses can protect against the negative effects of mitochondrial toxins. For example, exercise and calorie restriction in Caenorhabditis elegans promote mitochondrial maintenance and protect against the lethal effects from acute exposure to mitotoxicants such as rotenone and arsenic [54]. Also, food containing various phytochemicals have protective effects against injury caused by environmental toxins [5556].
More importantly, mitohormesis activation can be a fundamental mechanism to protect against mitochondrial dysfunction induced by multiple factors including inherited mitochondrial DNA (mtDNA) mutation [57], psychological stress [58], or aging [59]. For example, in a genetically modified mouse model designed to accumulate mtDNA mutations at every mtDNA replication cycle, premature death and multiple aging signs including muscle and brain atrophy, exercise intolerance, whitening of hair, and kyphosis occurred [6061]. However, this progeroid phenotype was entirely avoided by 5 months of endurance exercise [62]. This suggested the potential for the reversal of mitochondrial functions and the downstream physiological consequences by behavioral factors. If genetically inherited mitochondrial dysfunction can be reversed with exercise, mitochondrial dysfunction due to chronic mitochondrial toxin exposure can be recovered as well.
Exercise and diet are well-known heathy behaviors, but it is difficult to encourage people to make such healthy changes in their lifestyle [63]. However, if the biological link between unavoidable low-dose environmental chemical mixtures and heathy behavior is scientifically explained and understood by the public, it can provide a strong motivation for action. Identifying methods to activate stress responses effectively and safely would be an interesting research agenda in the field of mitochondrial toxins.
CONCLUSIONS
Mitochondrial stress responses can be a unifying theme that explains both the non-linear response of the negative effects caused by persistent mitochondrial toxins in the low-dose range and the benefits of exercise, calorie restriction (especially glucose), cognitive stimulation, and increased phytochemical intake. As exposure to mitochondrial toxins is inevitable in modern society, the optimal and effective activation of stress responses through lifestyle modifications should be investigated as reasonable and necessary measures against the negative effects caused by chronic exposure to low levels of environmental chemicals.
ACKNOWLEDGMENTS
This work was supported by “The Environmental Health Action Program (grant number 2016001370002),” funded by the Korea Ministry of Environment of the Republic of Korea.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.