An Update on the Effect of Incretin-Based Therapies on β-Cell Function and Mass
Article information
Abstract
Type 2 diabetes mellitus (T2DM) is a multifactorial disease with a complex and progressive pathogenesis. The two primary mechanisms of T2DM pathogenesis are pancreatic β-cell dysfunction and insulin resistance. Pancreatic β-cell dysfunction is recognized to be a prerequisite for the development of T2DM. Therapeutic modalities that improve β-cell function are considered critical to T2DM management; however, blood glucose control remains a challenge for many patients due to suboptimal treatment efficacy and the progressive nature of T2DM. Incretin-based therapies are now the most frequently prescribed antidiabetic drugs in Korea. Incretin-based therapies are a favorable class of drugs due to their ability to reduce blood glucose by targeting the incretin hormone system and, most notably, their potential to improve pancreatic β-cell function. This review outlines the current understanding of the incretin hormone system in T2DM and summarizes recent updates on the effect of incretin-based therapies on β-cell function and β-cell mass in animals and humans.
INTRODUCTION
Type 2 diabetes mellitus (T2DM) is a complex, progressive multi-factorial disease that is associated with various modes of pathogenesis and serious microvascular and macrovascular complications. Multiple classes of antidiabetic drugs designed to control glycemia in patients with T2DM are currently available and additional treatments continue to be developed. Although, multiple pathogenic processes contribute to T2DM [1], impaired insulin secretion and insulin resistance are recognized as the primary mechanisms underlying the pathophysiology of this disease.
Impaired pancreatic β-cell function is a prerequisite for the development of hyperglycemia and T2DM. Findings from the U.K. Prospective Diabetes Study [2] and A Diabetes Outcome Progression Trial [3] studies suggested that the progressive loss of β-cell function is associated with worsening hyperglycemia. Therefore, there is an urgent need for antidiabetic agents that improve, preserve or replace β-cell function.
Among the currently available antidiabetic drugs, the incretin-based therapies, glucagon-like peptide-1 analogue receptor agonists (GLP-1 RAs) and dipeptidyl peptidase-4 inhibitors (DPP4Is), are widely used antidiabetic drugs that target β-cell function and mass, as well as other systems that contribute to T2DM.
Here, we provide an update on our previous review of incretin hormones and their association with pancreatic β-cell function and mass in diabetes mellitus [4] based on recent experimental and clinical findings.
THE PHYSIOLOGICAL ROLE OF INCRETIN ON β-CELL FUNCTION AND MASS
In 1902, Bayliss and Starling first discovered 'secretin,' a chemical induced by food intake that is secreted from the intestinal mucosa and stimulates the secretion of hormones from the pancreas. In 1924, the glucose-lowering components of secretin were purified from gut extracts and renamed incretins (intestinal secretion of insulin) [5]. Oral glucose administration induces a much stronger insulin response compared with the intravenous (IV) administration of glucose, a phenomenon referred to as the 'incretin effect' [6], as it is associated with the release of incretins from the gut following the ingestion of glucose and nutrients. Two gut hormones, gastric inhibitory polypeptide (GIP; now referred to as glucose-dependent insulinotropic polypeptide) and GLP-1 are endogenous incretins that potentiate glucose-stimulated insulin secretion (GSIS) [7]. GIP is a 42-amino-acid hormone secreted from K cells in the upper small intestine, and GLP-1 is a 31-amino-acid hormone produced from a proglucagon precursor secreted from L cells in the lower intestine and colon. GLP-1 and GIP account for 50% to 70% of postprandial insulin secretion from pancreatic β-cells. After they are secreted, GLP-1 and GIP are rapidly degraded by the ubiquitous enzyme DPP4, which inactivates incretins by cleaving their 2 N-terminal residues.
Direct insulinotropic effects of incretin
Insulin secretion stimulated by IV glucose infusion is biphasic in non-diabetic subjects with a rapid, early peak (first-phase) followed by a second slower and gradually rising peak (second-phase) [8]. First-phase insulin secretion is rapidly stimulated by an increase in cytosolic Ca2+ and primarily results from the exocytosis of primed insulin granules. The slower second phase of insulin secretion is activated by cytosolic Ca2+, adenosine triphosphate (ATP) and cyclic adenosine monophosphate (cAMP) production, and results from the secretion of new insulin granules. GSIS can be regulated by amino acids, free fatty acids (FFAs), and non-nutrient secretagogues, such as incretin hormones, growth factors, and neurotransmitters. The first phase of insulin secretion plays a pivotal role in the transition from the fasting to the postprandial state by suppressing hepatic glucose production and lipolysis, and crossing the endothelial barrier of target cells to prime them for the effects of insulin. Similar to the first phase, second-phase insulin secretion inhibits hepatic glucose production, albeit to a lesser extent [9]. More importantly, second phase insulin secretion effects on increasing glucose utilization in peripheral tissues [10]. Although second-phase insulin secretion plays a critical role in glucose homoeostasis, its significance has been underestimated compared with the first phase.
GLP-1 and GIP directly increase GSIS in healthy subjects after glucose infusion, and the insulinotropic effects of GLP-1 and GIP are similar and additive [11]. The glucose-dependent activity of incretins has been demonstrated in multiple studies, and evidence suggests that a threshold glucose concentration might be required to promote incretin activity [12].
Both GIP and GLP-1 exert their effects by binding to their cognate receptors, the GIP receptor (GIPR) and the GLP-1 receptor (GLP-1R), respectively, which belong to the G-protein-coupled receptor (GPR) family. Receptor binding activates adenylate cyclase, thereby increasing levels of intracellular cAMP in pancreatic β-cells and, consequently, stimulating glucose-dependent insulin secretion [7]. Briefly, incretin-bound receptors increase intracellular cAMP levels, thereby activating protein kinase A (PKA) [13] and cAMP-activated guanine nucleotide exchange factors that target Ras-like GTPases 2 (Epac2; also referred to as cAMP-GEF-II) [14]. PKA and Epac2 mediate changes in ion-channel activity, and enhance cytosolic calcium levels and the exocytosis of insulin-containing granules. Together, these events contribute to the stimulation of insulin secretion in a glucose-dependent manner [7]. Activated PKA and Epac2 potentiate the effects of glucose. Activated PKA phosphorylates KATP channels, thereby facilitating channel closure and membrane depolarization. PKA and phosphatidylinositide 3-kinases (PI3K) inhibit Kv channels, which results in the prolongation of action potentials. Depolarization opens the voltage-gated Ca2+ channels, thereby increasing cytosolic Ca2+ levels and Ca2+ mobilization from intracellular stores via PKA- and Epac2-dependent mechanisms. The increase in Ca2+ levels triggers the fusion of insulin-containing granules with the plasma membrane and promotes insulin secretion from β-cells. Incretins promote the first phase of GSIS via these aforementioned pathways. In the second phase of insulin secretion, Ca2+ mobilization stimulates ATP synthesis in mitochondria, which further enhances membrane depolarization by promoting the closure of KATP channels, and activated Epac2 increases the density of insulin-containing granules near the plasma membrane, thereby facilitating insulin secretion from β-cells.
Indirect insulinotropic effects of incretin
The effects of GLP-1 on the central and peripheral nervous system have also been implicated in glucose homoeostasis [15]. As DPP4 directly cleaves and inactivates GLP-1 in the gut, less than 25% of the active metabolite of GLP-1 reaches the pancreatic islets. However, the effect of GLP-1 on afferent nerves in the intestinal mucosa and portal veins is still considered an important mechanism mediating insulin secretion [16].
GLP-1 is secreted by the intestine into the mesenteric capillaries in response to the ingestion of glucose and lipids and is subsequently released into the hepatic portal vein. These events activate the terminal ends of the vagus nerve to generate a neural signal directed towards the brain stem [17]. The solitary tract nuclei (nucleus tractus solitarii, NTS) and area postrema (AP) in the brain stem contain the cell bodies of neurons that synthesize GLP-1 and project their axons to the hypothalamus. Consequently, GLP-1 is secreted from the NTS and AP into the hypothalamus where it activates multiple receptors. A new signal sent to peripheral tissues via the autonomic nervous system regulates multiple functions and enhances GSIS. GLP-1 receptors have also been identified in the nodose ganglion of the afferent vagal nerve, and the portal infusion of GLP-1 in rats activates both the hepatic vagal afferent and the pancreatic vagal efferent pathways. In mice, inhibition of the sensory afferent pathway abolishes GLP-1 RA-mediated insulin secretion [18]. Furthermore, GLP-1 signaling in the central nervous system under hyperglycemic conditions promotes insulin secretion and activates the peripheral neural pathways that inhibit glucose uptake in muscle and promote hepatic glycogen storage [19]. GLP-1 activity might also mediate GLP-1 neuronal secretion in the brain in response to the detection of glucose by intestinal cells; however, the molecular pathways underlying this mechanism remain unclear.
Effects of incretin on β-cell mass
Both GIP and GLP-1 have demonstrated non-insulinotropic actions, such as controlling β-cell proliferation and survival [7,20]. GIP has also exhibited an anti-apoptotic function in pancreatic β-cells that is mediated by the activation of the cAMP response element-binding protein (CREB) and protein kinase B (Akt/PKB) pathways. When activated, these two pathways promote the transcription of the anti-apoptotic gene Bcl-2 and downregulate the proapoptotic gene BAX, thereby promoting β-cell apoptosis in response to glucolipotoxicity. Furthermore, GIP suppresses the mitochondria-mediated apoptotic pathway by activating Akt/PKB in INS-1 cells. Suppression of p38 mitogen-activated protein kinases (MAPK) and JNK are critical for the anti-apoptotic functions of GIP in INS-1 cells exposed to endoplasmic reticulum (ER) stress and genotoxic stress [21].
The activation of GLP-1R by exendin-4 inhibits apoptosis in MIN6 cells exposed to hydrogen peroxide in a cAMP- and PI3K-dependent manner, upregulates Bcl-2 and Bcl-xL, and downregulates poly(ADP-ribose) polymerase. Activation of PKA and Epac by elevated cAMP levels inhibits caspase-3 activation and apoptosis in palmitate-treated RINm5F cells. Activation of Akt/PKB, a downstream effector of PI3K, by GLP-1 prevents the apoptosis of INS-1 cells in response to glucolipotoxicity or staurosporine, a rapid activator of the mitochondrial apoptotic pathway. The anti-apoptotic function of activated Akt/PKB is potentially mediated by the activation of nuclear factor-κB (NF-κB) and the upregulation of the anti-apoptotic NF-κB target genes Bcl-2 and IAP2. GLP-1R activation also attenuates the ER stress response, thereby promoting β-cell survival in INS-1 cells and in human islets. A critical distinction of the anti-apoptotic function of GLP-1 compared with GIP is the requirement of PI3K. However, the physiological impact of this difference remains unclear, and further investigation into the effects underlying the anti-apoptotic functions of GIP and GLP-1 is needed to identify potential therapeutic targets that might increase β-cell mass by inhibiting apoptosis.
Another important effect of GIP and GLP-1 activity on β-cell mass is the stimulation of β-cells and/or progenitor cell proliferation [7,20]. Activation of GIPR induces β-cell proliferation. GIP activates the Raf-Mek1/2-ERK1/2 signaling pathway via cAMP/PKA signaling in GIPR over-expressing CHO cells. Constitutive GLP-1R activation increases β-cell proliferation and neogenesis in non-diabetic and diabetic rodents. By activating the cAMP/PKA/CREB, PI3K, and ERK1/2 pathways, GLP-1 is considered a growth and differentiation factor for mature β-cells and β-cell progenitors. GIP and GLP-1 induce the transcription of cyclin D1, a molecule critical for cell cycle progression from the first gap phase (G1) to the synthesis (S) phase in most cell types. GLP-1 also replenishes insulin stores and prevents β-cell exhaustion by upregulating insulin at the mRNA and protein levels via activation of PKA-dependent and -independent signaling pathways. The mobilization of Ca2+ elicited by the cAMP/PKA pathway activates Ca2+/calcineurin/nuclear factor of activated T cells (NFAT). NFAT is a key mediator of cAMP/PKA pathway-mediated of the gene encoding insulin and of β-cell growth and function. However, inhibitory regulatory mechanisms limit GLP-1-induced β-cell proliferation [22].
PANCREATIC β-CELL FUNCTION AND MASS IN TYPE 2 DIABETES MELLITUS
β-Cell dysfunction and loss in type 2 diabetes
First-phase insulin secretion is abolished in T2DM [23]. Although hyperglycemia might contribute to defects in insulin secretion, these defects persist in T2DM patients after glycemic control is achieved [24]; therefore, it has been suggested that patients with T2DM have an intrinsic β-cell defect. A reduction in first-phase insulin secretion has been observed not only in patients with T2DM but also in patients with symptoms of prediabetes [25]. Thus, β-cell abnormalities might precede the development of T2DM, and first-phase insulin secretion has been proposed to be a prognostic marker of T2DM in high-risk populations [26]. Although the slow, gradual second phase of GSIS that follows first-phase secretion is reduced and delayed in T2DM, it remains partially functional and is sometimes exaggerated due to the hyperglycemia induced by defective first-phase insulin secretion [23].
Butler et al. [27] reported that obese and lean T2DM patients exhibited a reduction in relative β-cell volume of 63% and 41%, respectively, compared with non-diabetic obese and lean individuals, respectively. In addition, they observed a 40% reduction in β-cell volume in obese individuals with IFG compared with non-diabetic obese individuals. The study concluded that the decrease in β-cell mass observed in patients with T2DM is mediated by an increase in β-cell apoptosis. Yoon et al. [28] and Rahier et al. [29] later reported that β-cell mass in patients with T2DM was reduced compared with matched controls. The progressive decline in β-cell mass associated with disease duration might contribute to the progressive deterioration of glucose homoeostasis and further impair insulin secretion.
However, fasting hyperglycemia only develops in patients with >90% loss in β-cell mass [30]. Furthermore, despite the 50% reduction in β-cell mass in patients with T2DM, maximum insulin secretion in response to IV glucose and arginine is only 15% of that observed in non-diabetic individuals [31]. Thus, it is unlikely that the reduction in β-cell mass alone can account for the severity of the intrinsic defects in the secretory machinery observed in patients with T2DM.
Cellular and molecular pathways involved in β-cell dysfunction in T2DM
Multiple factors, including glucotoxicity, lipotoxicity, inflammation, cytokines, islet amyloid deposits, autoimmunity, incretin defects, and insulin resistance, have been suggested to be key pathophysiological features of defective β-cells in patients with T2DM (Fig. 1) [4].
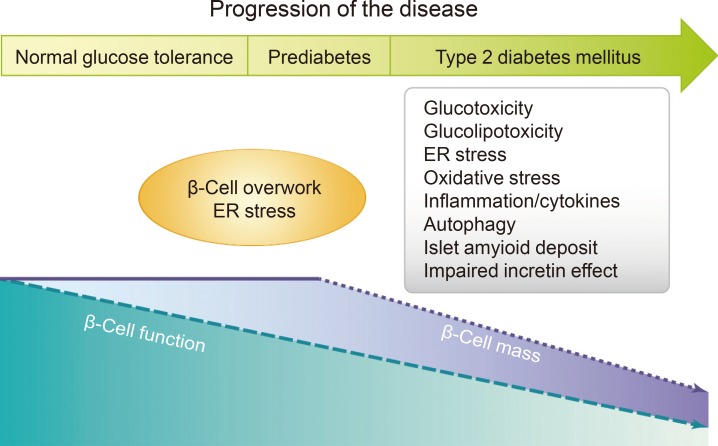
β-Cell function and mass changes over the time course of type 2 diabetes mellitus and proposed mechanisms. ER, endoplasmic reticulum. Figure extracted from: Chon S, Riveline JP, Blondeau B, Gautier JF. Incretin-based therapy and pancreatic beta cells. Diabetes Metab 2014;40:411-22 [4]. Copyright ©2014. With permission of Elsevier Masson SAS.
Glucotoxicity is defined as β-cell failure induced by the chronic hyperglycemia that results from reductions in insulin secretion and insulin gene expression, and it is clearly distinct from β-cell exhaustion [32]. Lipotoxicity or 'glucolipotoxicity' refers to the negative effect of chronically elevated plasma FFA levels on β-cell function [33,34]. A number of mechanisms have been proposed to account for glucotoxicity-mediated β-cell dysfunction and death, including ER stress, oxidative stress, islet cell inflammation, and mitochondrial dysfunction. In addition, several mechanisms have been implicated in lipotoxicity (or glucolipotoxicity), including ceramide formation, oxidative stress, inflammation, and, more recently suggested, ER stress, and autophagy. Recent studies have demonstrated that autophagy, a lysosome-mediated catabolic process implicated in the etiology of various age-related diseases, is also associated with lipotoxicity and diabetes. In addition, islet amyloid deposition by islet amyloid polypeptide, a 37-amino-acid polypeptide co-secreted with insulin by β-cells, has been suggested to contribute to β-cell dysfunction and death.
Defective incretin effect in type 2 diabetes mellitus
It is well-established that the incretin effect is impaired in T2DM patients, contributing approximately only 20% and 35% of the response to 50 and 75 g of oral glucose, respectively [35]. Defects in the incretin effect in patients with T2DM are suggested to be an early phenomenon in the development of T2DM, rather than an event that precedes the onset of the disease.
Although modest reductions in GLP-1 levels have been observed in patients with T2DM with suboptimal glycemic control and a relatively long disease duration [36], most studies have reported that levels of GLP-1 secretion in response to oral glucose were similar in T2DM patients and non-diabetic subjects [37]. The results of studies evaluating GIP secretion in patients with T2DM have been inconsistent, and there are reports of similar, increased or decreased levels of GIP secretion compared with non-diabetic controls. These findings suggest that there are no significant defects in GIP secretion in patients with T2DM.
A reduction in the insulinotropic action of GIP has been observed in patients withT2DM [35]. This defect appears to be associated with diabetes pathophysiology, as it has not been observed in relatives of patients with T2DM or in women with a history of gestational diabetes, but has been observed in patients with diabetes induced by chronic pancreatitis. Moreover, a reduction in the insulinotropic effect of GIP has been observed in experimental states of insulin resistance induced by a high-fat diet, sedentary lifestyle and steroid use [38]. GIPR downregulation in conjunction with poor β-cell recognition, defective receptor signaling and general β-cell dysfunction have all been proposed as mechanisms underlying the reduction in GIP activity in patients with T2DM [35]. Hyperglycemia might be another important contributing factor, as improvements in glycemia in T2DM patients to nearly normal levels has been shown to improve the insulinotropic effect of GIP.
The insulinotropic activity of GLP-1 is partially preserved, albeit reduced, in patients with either T2DM or impaired glucose tolerance and in first-degree relatives of patients with T2DM [35]. Notably, a reduction in GLP-1-induced insulin secretion has also been observed under conditions of experimental insulin resistance induced by a sedentary lifestyle, a high-fat diet and steroid treatment [38], suggesting that insulin resistance alone can impair the insulinotropic activity of GLP-1. Thus, impaired GLP-1-induced insulin secretion might be a very early event in the development of T2DM. Several mechanisms have been proposed to that might mediate the reduction in the GSIS activity of GLP-1 have been proposed [35]. First, this defect might be a consequence of general β-cell dysfunction in T2DM. Second, the pathogenic features of diabetes, including hyperglycemia, hyperlipidemia, and insulin resistance, might disrupt GLP-1 activity, as the achievement of near normal levels of blood glucose and the administration of lipid-lowering agents improved GLP-1-induced GSIS in mouse models. Third, some patients with T2DM might have genuine GLP-1 resistance due to genetic aberrations that disrupt the ability of GLP-1 to signal to β-cells. Examples of such genetic aberrations include a polymorphism in the transcription factor 7-like 2 (TCF7L2) gene that impairs GLP-1-induced insulin secretion and the genetic variant of the Wolfram syndrome 1 (WFS1) gene that disrupts GLP-1-induced insulin secretion but not IV glucose-induced insulin secretion.
EFFECTS OF INCRETIN-BASED THERAPY ON β-CELL FUNCTION AND MASS IN PRECLINICAL STUDIES
The effect of GLP-1 is preserved in T2DM, but the incretin effect of GIP is absent even when administered in supraphysiological doses. Therefore, therapeutic approaches based on targeting the incretin effect in T2DM have primarily focused on GLP-1. Both native GLP-1 and GIP are rapidly degraded by DPP4; therefore, restoring normal GLP-1 and GIP levels would require continuous parenteral administration. To overcome this limitation, two pharmacological approaches have been developed: (1) the use of DPP4-resistant GLP-1 RAs with a prolonged half-life that provide supraphysiological levels of GLP-1; (2) the use of DPP4Is to protect incretins from DPP4-mediated degradation and increase endogenous incretin hormone levels.
GLP-1 receptor agonists
Currently available GLP-1 RAs include the once-daily therapies exenatide, liraglutide, lixisenatide, and the once-weekly long-acting slow-release GLP-1 RAs exenatide, albiglutide, and dulaglutide [39]. Exenatide (synthetic exendin-4), which is derived from the saliva of the Gila monster, exhibits 53% homology to native GLP-1. Due to its structural similarity to native GLP-1, exenatide is able to bind to the GLP-1 receptor in vivo; thereby, stimulating glucose-dependent insulin secretion. Liraglutide, with 97% homology, is even more similar to native GLP-1 compared with exenatide. Liraglutide's prolonged half-life of 13.1 hours is due to its delayed absorption and strong resistance against DPP4 degradation, which primarily result from a fatty acid substitution that promotes albumin bonding. Lixisenatide is a synthetic version of exendin-4 and it is resistant to degradation by DPP4 as a result of a C-terminal modification in which six lysine residues are added and one proline residue is deleted [40]. Once-weekly exenatide is released into the circulation via poly-microsphere delivery over a period of 10 weeks and exhibits a chemical composition identical to once-daily exenatide. Albiglutide has an extended duration of action, with a half-life of 5 days, as a result of the addition of two amino acids that bind to albumin in vivo. Dulaglutide's extended duration of action is due to amino acid modifications that promote resistance to DPP4 degradation, as well as to the large size of the molecule which results in reduced renal clearance.
A large body of preclinical data has demonstrated that the enhancement of GLP-1R signaling by either GLP-1 RAs or DPP4Is enhances β-cell function, proliferation and regeneration and diminishes β-cell apoptosis; thereby, providing a net increase in functional β-cell mass [20]. Exenatide enhances glucose-dependent first- and second-phase insulin secretion via the GLP-1R downstream signaling pathway, increases β-cell mass via β-cell neogenesis and proliferation, and reduces apoptosis [11]. Exendin-4 was reported to promote β-cell proliferation via the PI3K/Akt signaling pathway, rather than the ERK/MAPK pathway, in vivo and in vitro [41]. Exendin-4 promoted the transcription of the Neurod 1 and Glut2 genes, and induced the differentiation of mouse embryonic stem cells into endocrine and insulin-producing cells [42]. In addition, exendin-4 efficiently improved blood glucose levels and glucose tolerance in β-cell-specific Atg7-deficient mice primarily by enhancing insulin secretion, reducing apoptosis and increasing proliferation [43]. Although the precise mechanism underlying the efficacy of exendin-4 requires further investigation, these findings suggest that exendin-4 might effectively target β-cell dysfunction and prevent the increase in β-cell apoptosis that is associated with autophagy deficiency without altering the cellular autophagy machinery.
Liraglutide also increases β-cell proliferation and β-cell mass in ob/ob mice [44]. Studies using primary neonatal rat islets demonstrated that liraglutide inhibits both cytokine- and FFA-induced apoptosis via the PI3K-mediated pathway [45]. In addition, liraglutide increases β-cell mass, not only by directly regulating cell proliferation, differentiation, and apoptosis but also by suppressing the oxidative and ER stress that results from the amelioration of glucotoxicity [46]. Furthermore, GLP-1R signaling enhances β-cell survival in human islets in vitro and in islet transplant studies of rodent and human islets in animals in vivo [47].
Recent reports revealed that sustained liraglutide treatment in normoglycemic mice is associated with increased insulin secretion from isolated islets in vitro, even in the absence of direct GLP-1 RA stimulation, as well as increased insulin sensitivity [48]. In that study, both β-cell and α-cell mass decreased and the α/β-cell ratio remain unchanged after 1 week of liraglutide treatment; however, ductal cell proliferation increased. This suggests that GLP-1 RA increases insulin sensitivity and enhances insulin secretion from existing β-cells under conditions of normal glycemia; however, the effects on the proliferation of exocrine pancreatic cells have yet to be determined. Liraglutide has also been shown to prevent β-cell dysfunction in rats under conditions of starvation followed by excessive feeding [49]. The findings of this study suggested that the clinical benefits of liraglutide are associated with an increase in β-cell mass due to increased β-cell proliferation, decreased apoptosis and enhanced insulin secretion.
Conversely, a recent study suggested that pharmacological GLP-1R activation with exendin-4 or liraglutide induces protein synthesis; thereby, increasing pancreatic mass independent of changes in DNA content or cell proliferation in mice [50]. In this study, GLP-1R–dependent upregulation of Reg family members and proteins important for protein translation and export, including Fam129a, eIF4a1, Wars, and Dmbt1, were implicated in the increase in pancreatic mass. Although the cellular sites of GLP-1R expression that are required for the transduction of these signals to the exocrine pancreas remain unclear, these data suggest a potential mechanism underlying GLP-1R signaling to the exocrine pancreas. In addition, these data provide a potential explanation for previous observations that the pharmacological administration of GLP-1R agonists increased pancreatic mass in preclinical studies.
Lixisenatide has been shown to restore normal blood glucose levels and human plasma insulin levels and to improve glucose tolerance compared with control treatment in diabetic human islet-engrafted immunodeficient mice [51]. However, the proportions of proliferating and apoptotic β-cells at graft recovery were not different between the two groups. Nevertheless, there was a significant 2- to 3-fold increase in human β-cells in lixisenatide-treated mice, suggesting that increased β-cell survival coupled with enhanced β-cell function induced by lixisenatide treatment might provide the greatest therapeutic benefits to diabetic patients with a reduction in functional islets, as the proliferative capacity of human β-cells is limited [51].
However, the proliferative-enhancing actions of GLP-1 RA are markedly attenuated or absent in older rodents, most likely reflecting the epigenetic alterations that impair β-cell mitosis, the down-regulation of p27 and the increased expression of p16INK4A, which is a negative regulator of cyclin-dependent kinase. The capacity of GLP-1 RA to enhance proliferation in human islets from older donors remains unclear, but it appears to be limited [52].
DPP4 inhibitors
DPP4Is are orally administered small molecule drugs that compete with DPP4 substrates for the active sites in the enzyme and that inhibit >80% of DPP4 activity. Thus, DPP4Is increase circulating levels of endogenous active GLP-1 and GIP by approximately 2- to 3-fold and exert a glucose-dependent dual action on both α- and β-cell function by which they stimulate insulin secretion and suppress glucagon secretion under hyperglycemic conditions.
Currently, multiple DPP4Is are approved for use in the treatment of T2DM, including sitagliptin, vildagliptin, saxagliptin, linagliptin, anagliptin, alogliptin, gemigliptin. In addition, a number of other DPP4Is are pending approval. The various DPP4Is slightly differ in their structure, absorption rate, distribution, metabolism, and elimination, as well as in their potency and duration of action [53].
DPP4I treatment in rodent models of diabetes improved fasting and non-fasting glucose, enhanced plasma insulin levels, reduced plasma glucagon levels and increased pancreatic insulin stores [54]. However, these studies were conducted in different rodent models and used different methods to evaluate glucose metabolism and pancreatic function, factors that preclude direct comparisons of the studies. The antihyperglycemic effect of DPP4I in dual incretin receptor knockout mice has been reported to require functional incretin receptors on islet cells [55].
Several studies have assessed the effects of acute and chronic treatment with DPP4I on pancreatic islet and β-cell morphology in rodents [54]. Chronic DPP4I treatment for 2 to 3 months increased β-cell mass by promoting cell proliferation and reducing apoptosis. Interestingly, after a 12-day drug washout period, the beneficial effects on β-cell mass were maintained in neonatal rats treated with a DPP4I for 19 days. In contrast, other studies reported that DPP4I treatment had no effect on total β-cell mass; however, some studies demonstrated that DPP4Is exerted a beneficial effect on the intra-islet distribution pattern of α- and β-cells. A recent study reported that vildagliptin treatment increased β-cell mass by indirectly reducing cell apoptosis, oxidative stress and ER stress, and directly regulating cell differentiation and proliferation in diabetic mice [56]. Vildagliptin prevented islet inflammation in a high fat diet-induced obesity mouse model of advanced age [57]. Furthermore, vildagliptin promoted β-cell survival and downregulated ER stress markers in db/db mice [58]. Linagliptin exerts a protective effect on β-cell turnover and function under diabetic conditions, (gluco-, lipo-, and cytokine toxicity), via an anti-inflammatory/antioxidant pathway and the stabilization of GLP-1 [59].
DPP4 inhibition exerted durable effects on pancreatic islet mass and/or insulin content, an effect that was not observed with sulfonylurea. Furthermore, combination treatment with a DPP4I and either a thiazolidinedione (TZD) or an α-glucosidase inhibitor increased pancreatic insulin levels compared with either agent alone. Sitagliptin alone or in combination with metformin preserved β-cell mass by inhibiting apoptosis and increasing proliferation in transgenic rats overexpressing humanislet amyloid polypeptide in β-cells [60]. However, sitagliptin treatment was associated with increased pancreatic ductal turnover and ductal metaplasia.
One study evaluated the morphology and function of islets in a mouse model of β-cell injury/regeneration treated with a DPP4I (MK-0626) and a GLP-1 RA (liraglutide), either alone or in combination [61]. A 2-week intervention in diphtheria toxin-injected mice altered islet morphology and function. MK-0626, but not liraglutide, enhanced β-cell proliferation and GSIS, whereas liraglutide, but not MK-0626reduced α-cell mass. The pro-proliferative effect of MK-0626 was abolished by the co-administration of the GLP-1 RA exendin-(9-39). A comparison of the effects of DPP4I and GLP-1 RA treatment in this mouse model demonstrated that the two anti-diabetic drugs have distinct effects on islet morphology and function.
EFFECTS OF INCRETIN-BASED THERAPY ON β-CELL FUNCTION AND MASS IN CLINICAL STUDIES
Effects on β-cell function
GLP-1 receptor agonists
Exenatide and liraglutide exert beneficial effects on β-cell function in patients with T2DM. Exenatide improved β-cell function in both static and dynamic tests, as measured by the homoeostasis model assessment of β-cell function (HOMA-B) and by glucose- and arginine-stimulated C-peptide secretion in hyperglycemic clamp assays in clinical studies [62]. Short-term IV exenatide treatment for 3 hours normalized both first- and second-phase insulin secretion following IV glucose challenge in patients with T2DM [20]. Bunck et al. [63] evaluated the long-term effects (1 year of administration) of exenatide in T2DM patients treated with metformin. Exenatide treatment improved β-cell function compared with insulin glargine, with improvements in first- and second-phase glucose-stimulated C-peptide and arginine-stimulated C-peptide under conditions of hyperglycemia. However, these benefits were lost in both groups 4 weeks after the drug was discontinued.
Chang et al. [64] reported that a single injection of liraglutide restored β-cell responsiveness to a graded glucose infusion in a study evaluating the effects of different doses of liraglutide on first- and second-phase insulin secretion compared with placebo in patients with T2DM [65]. The two highest doses of liraglutide improved first- and second-phase insulin secretion and arginine-stimulated insulin secretion during hyperglycemia. A meta-analysis of the Liraglutide Effect and Action in Diabetes (LEAD) trials 1, 2, and 5 revealed that liraglutide significantly improved β-cell function, as measured by HOMA-B, and the proinsulin/insulin (PI/I) ratio [66]. Lixisenatide improved both fasting and prandial glycemia, with a particularly pronounced effect on prandial blood glucose. In addition, lixisenatide significantly improved β-cell function, as assessed by HOMA-B [67]. Lixisenatide was also provided significantly greater reductions in postprandial insulin, C-peptide, and glucagon by slowing the rate of gastric emptying than liraglutide [68]. Dulaglutide has also been shown to increase β-cell function, as demonstrated by an increase in HOMA2-B, when administered as monotherapy or in combination with metformin with or without other antidiabetic agents [69].
Few clinical trials have evaluated the long-term effects of GLP-1 RAs on β-cell function and glycemic control in patients with T2DM. In an extended study reported by Bunck et al. [70], improvements in β-cell function, as assessed by an increase in disposition index, persisted in the exenatide group 4 weeks after drug discontinuation in a subgroup analysis of 217 patients during a 3-year follow-up period. However, it is unclear whether this difference directly reflected improvements in β-cell function or was an indirect consequence of the substantial 7.9 kg weight loss observed in subjects who received exenatide [47]. A recent study that compared ≤4.5 years of treatment with exenatide and glimepiride as add-ons to metformin reported that exenatide provided long-term improvements in glycemic control and disposition index. However, these effects might have been related to improvements in body weight and high drop out rate due to treatment failure [71]. Liraglutide treatment for 2 years provided greater improvements in glucose control compared with glimepiride in patients with T2DM; however, no formal assessment of β-cell function after drug discontinuation was reported. Regardless, approximately 20% of patients failed to maintain glycemic control with liraglutide [72].
Thus, current clinical study data do not definitively support the concept that GLP-1 RA treatment provides sustained improvements in β-cell mass and function over time in patients with T2DM.
DPP4 inhibitors
One study evaluated the effect of DPP4Is on β-cell function using static and dynamic tests in humans [54]. DPP4I monotherapy provided improvements in fasting measures of β-cell function, including HOMA-B and the PI/I ratio. Improvements in fasting parameters of β-cell function were also observed in patients receiving DPP4Is as an add-on therapy to oral antidiabetic drugs such as metformin, sulfonylurea and TZDs. DPP4Is also improved early β-cell response, also referred to as the insulinogenic index, in several trials when administered either as monotherapy or as add-on therapy. Both sitagliptin and vildagliptin improved postprandial insulin/glucose area under the curve (AUC) compared with placebo. DPP4Is also improved several model-derived parameters of β-cell function. Mari et al. [73] reported that vildagliptin improved model-derived insulin secretory rate (ISR) by 17% and β-cell glucose sensitivity by 40%. Furthermore, 12 weeks of sitagliptin treatment provided improvements in first- and second-phase and arginine-stimulated insulin secretion in clamp tests in patients with T2DM [74].
Nevertheless, there is little evidence supporting the long-term effects of DPP4Is on β-cell function in humans, although some randomized controlled trials have evaluated DPP4Is during an extension period of up to 2 years [54]. Sustained benefits in the PI/I ratio were observed in patients treated with vildagliptin in combination with metformin for 1 year, and sustained improvements in both PI/I ratio and HOMA-B were observed in patients treated with sitagliptin in combination with metformin for 1 year. In studies of DPP4I treatment as an add-on to metformin for 2 years, vildagliptin stabilized β-cell function, as assessed by ISR/G (ISR AUC0–2h/glucose AUC0–2h), and sitagliptin provided improvements in insulin/glucose AUC that persisted after a washout period of 4 to 7 days. However, in other studies evaluating patients treated with DPP4Is for 1 year, improvements in β-cell function assessed by meal tests were not maintained after a 4-week washout period [7576]. Another study evaluating β-cell function dynamic in patients treated with DPP4I for 1 year using intravenous glucose tolerance test reported that β-cell function parameters reverted to baseline values after a 12-week washout period [77]. The available data indicate that DPP4Is provide stable improvements in indicators of β-cell function after prolonged treatment of up to 2 years. A recent systematic review of DPP4Is evaluated glycemic durability as a surrogate marker of improvements in β-cell function [78]. However, this review reported that improvements in glycosylated hemoglobin (HbA1c) provided by DPP4Is in patients with T2DM significantly declined during the second year of treatment. A recent meta-analysis of large randomized clinical trials (RCTs) evaluating the effect of 2 years of saxagliptin treatment demonstrated that saxagliptin prevented further decline in β-cell function compared with placebo, especially in treatment-naive patients and patients with higher HOMA-2B at baseline [79]. Although this study initially aimed to assess cardiovascular outcomes, the results of this study of a relatively large population of 4,720 patients suggested that DPP4Is might attenuate the progression of T2DM and that DPP4Is might be more effective during the early stages of T2DM [79]. However, additional long term follow-up studies are needed to confirm that DPP4Is provide durable improvements in β-cell function.
Some studies have also evaluated the effects of DPP4Is on β-cell function in patients with recent-onset latent autoimmune diabetes in adults (LADA) [80]. One study reported that β-cell function was maintained in LADA patients treated with sitagliptin and insulin compared with patients treated with insulin alone, suggesting that the lack of decline in β-cell function in the sitagliptin group might be related to metabolic- or immune-mediated effects. However, additional studies with larger cohorts are needed to assess the long-term clinical outcomes associated with DPP4Is and to precisely characterize the mechanism underlying DPP4I-mediated β-cell preservation in patients with LADA.
Effects on β-cell mass
Butler et al. [81] reported that incretin-based therapies, primarily sitagliptin, increase β-cell mass and markedly expand the exocrine pancreas by increasing proliferation/dysplasia and α-cell hyperplasia and that they have the potential to promote the development of endocrine tumors, as evidenced by a limited histopathological study of organ donors with T2DM. This paper was criticized due to various issues with the methodologies used, including the nature of the control population [82], as well as because their findings could not be reproduced by two independent groups that evaluated the same samples [8384]. In addition, both the U.S. Food and Drug Administration and European Medicines Agency have stated that causal associations between incretin-based drugs and pancreatitis or pancreatic cancer are inconsistent with their extensive reassessment of preclinical and clinical data regarding incretin-based drugs [85]. Nonetheless, the potential dangers of these drugs on the pancreas should continue to be evaluated.
A case study of a 40-year-old man with type 1 diabetes mellitus (T1DM) receiving exenatide reported that the patient experienced a change in HbA1c from 8.7% to 7.3% after 11 months of treatment and his basal insulin requirement decreased by 25% [86]. Exendin-4 treatment in patients with T1DM reduced postprandial glycemia by reducing glucagon levels [87]. However, another study evaluating the use of exenatide for 6 to 9 months in patients with long-standing T1DM reported that no effects on residual β-cell function were observed [88]. A recent RCT evaluating liraglutide treatment in patients with T1DM taking insulin reported that liraglutide did not significantly affect HbA1c compared with placebo after 24 weeks of treatment, although liraglutide was associated with reductions in hypoglycemic events, bolus, and total insulin dose and body-weight, and an increase in heart rate [89].
This result suggested that the ideal candidates for incretin-based therapy in T1DM might be younger patients and patients at an earlier stage of the disease, when significant β-cell mass still exists. However, a recent small open-label study reported that the addition of incretin-based therapies to the treatment regimen of patients with new-onset T1DM decreased daily insulin requirements but had no significant effect on endogenous insulin secretion [90].
The effect of GLP-1 RA treatment administered at the time of islet transplantation to preserve islet mass and function in patients with T1DM has also been investigated. Exenatide treatment in patients undergoing islet transplantation significantly stabilized glycemic control and transplanted islet function [9192]. However, after exenatide treatment was discontinued, the improvements in β-cell function were not maintained. The Edmonton protocol with exenatide and etanercept after allogenic islet transplantation (1 to 3 times) for 15 months in patients with C-peptide-negative T1DM improved islet graft function and facilitated insulin independence with fewer islets compared with the small number of control patients who received the Edmonton protocol alone [93]. Regarding the mechanism underlying this effect, a recent study evaluating human islets from multiorgan donors demonstrated that exenatide exerted anti-inflammatory and cytoprotective effects on human islets in vitro and in immunodeficient mice after transplantation and that these effects were mediated by the upregulation of serine protease inhibitor 9, a protein with critical immunoregulatory properties [94].
The effect of DPP4Is on β-cell mass in humans has not been evaluated in clinical studies. One study evaluated the effect of sitagliptin treatment in patients with newly diagnosed T1DM patients who had undergone autologous non-myeloablative hematopoietic stem cell transplantation [95]. Two of the patients in the study who received sitagliptin (100 mg/day) exhibited a significant increase in C-peptide levels, and insulin replacement therapy was discontinued 2 months after the transplantation. Both patients did not require exogenous insulin for an additional 6 months. This result suggests that DPP4Is might promote the differentiation of transplanted stem cells and play an immunoregulatory role in autoimmune insulitis [96].
In contrast to what has been observed in rodent studies, there is currently no clear evidence that incretin-based therapies increase β-cell mass in humans. This discrepancy might be due to a number of differences between humans and rodents, including intrinsic species differences, differences in β-cell turnover, different responses to regenerative stimuli, and differences in the amount of drug administered and the duration of treatment [9798].
More accurate and non-invasive methods to assess functional β-cell mass in vivo are needed to facilitate the evaluation of any treatment on β-cell mass in humans [99]. Furthermore, additional long-term RCTs evaluating relatively larger numbers of patients, especially patients with new-onset T1DM and at different stages of preclinical T1DM, are needed to confirm the effects of GLP-1 RAs and DPP4Is on β-cell mass.
CONCLUSIONS
Pancreatic β-cell defects are the critical prerequisite for the development of T2DM. The results of recent investigations into β-cell defects in diabetes suggest that glucotoxicity and glucolipotoxicity most likely decrease β-cell function by multiple mechanisms, including ER stress, oxidative stress, islet inflammation, mitochondrial dysfunction, ceramide formation, and autophagy. Antidiabetic treatments aiming to preserve, or even increase, β-cell function/mass are considered critical to long-term glucose control. Incretin-based therapies have been a focus of interest due to their potential effects on β-cells (Fig. 2).
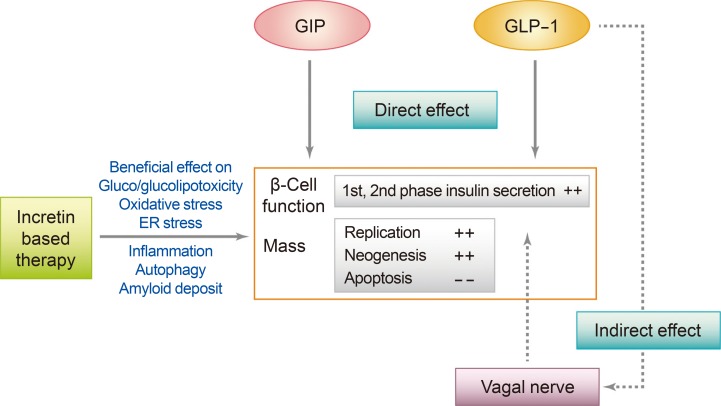
Effect of incretins and incretin based therapy on pancreatic β-cell in animal models. GIP, gastric inhibitory polypeptide; GLP-1, glucagon-like peptide-1; ER, endoplasmic reticulum. Figure extracted from: Chon S, Riveline JP, Blondeau B, Gautier JF. Incretin-based therapy and pancreatic beta cells. Diabetes Metab 2014;40:411-22 [4]. Copyright ©2014. With permission of Elsevier Masson SAS.
Various new incretin-based approaches aiming to improve β-cell function/mass are being evaluated. GPR40 is a Gq-coupled receptor for FFAs that is predominantly expressed in pancreatic β-cells, and GPR40 agonists have been investigated as new therapeutic agents in T2DM. In a recent study, the addition of a novel small molecule GPR40 agonist to a GLP-1-based therapy potentiated insulin secretion and improved glucose intolerance in isolated islets in mice and in a diabetic rat model [100]. Another novel therapeutic agent, the GLP-1 gastrin dual agonist ZP3022, provided sustained improvements in glycemic control that were accompanied by an increase in β-cell mass, β-cell proliferation, and mean islet mass compared with liraglutide alone in mice [101]. In addition, sitagliptin treatment in combination with a GPR119 agonist, a drug that promotes the accumulation of intracellular cAMP and enhances GLP-1 and insulin secretion, significantly increased plasma levels of active GLP-1, improved glucose clearance, stimulated both α- and β-cell replication, and augmented β-cell mass in diabetic mice [102].
To date, there is a lack of sufficient evidence to confirm that incretin-based therapies can modify the progressive decline in β-cell function in T2DM in humans. It is also unclear if the beneficial effects of incretin-based therapy on β-cell mass observed in animal studies are applicable to humans. Although, further studies should be conducted to evaluate the long-term effects of incretin-based therapy on β-cell function, glucose control, and safety issues associated with their effects on cell proliferation, a growing body of evidence suggests that incretin-based therapies have the potential to modify the natural course of diabetes.
ACKNOWLEDGMENTS
This review was supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2012S1A2A1A01031559) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI14C2700).
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.