Reducing Oxidative Stress and Inflammation by Pyruvate Dehydrogenase Kinase 4 Inhibition Is Important in Prevention of Renal Ischemia-Reperfusion Injury in Diabetic Mice
Article information

Abstract
Background
Reactive oxygen species (ROS) and inflammation are reported to have a fundamental role in the pathogenesis of ischemia-reperfusion (IR) injury, a leading cause of acute kidney injury. The present study investigated the role of pyruvate dehydrogenase kinase 4 (PDK4) in ROS production and inflammation following IR injury.
Methods
We used a streptozotocin-induced diabetic C57BL6/J mouse model, which was subjected to IR by clamping both renal pedicles. Cellular apoptosis and inflammatory markers were evaluated in NRK-52E cells and mouse primary tubular cells after hypoxia and reoxygenation using a hypoxia work station.
Results
Following IR injury in diabetic mice, the expression of PDK4, rather than the other PDK isoforms, was induced with a marked increase in pyruvate dehydrogenase E1α (PDHE1α) phosphorylation. This was accompanied by a pronounced ROS activation, as well as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β), and monocyte chemoattractant protein-1 (MCP-1) production. Notably, sodium dichloroacetate (DCA) attenuated renal IR injury-induced apoptosis which can be attributed to reducing PDK4 expression and PDHE1α phosphorylation levels. DCA or shPdk4 treatment reduced oxidative stress and decreased TNF-α, IL-6, IL-1β, and MCP-1 production after IR or hypoxia-reoxygenation injury.
Conclusion
PDK4 inhibition alleviated renal injury with decreased ROS production and inflammation, supporting a critical role for PDK4 in IR mediated damage. This result indicates another potential target for reno-protection during IR injury; accordingly, the role of PDK4 inhibition needs to be comprehensively elucidated in terms of mitochondrial function during renal IR injury.
Highlights
• PDK4 linked to ROS and inflammation in IR-induced kidney injury in diabetic mice.
• DCA and shPDK4 reduced ROS, inflammation, and kidney cell damage.
• This identifies PDK4 as a target for renal protection in diabetic mice with IR injury.
INTRODUCTION
Acute kidney injury (AKI) is a clinical condition defined by an abrupt decrease in kidney function accompanied by a fatal and rapid loss of the renal excretory function, which is represented by elevated serum creatinine and accumulation of uremic waste products [1]. AKI has multifactorial causes, including intrinsic (e.g., acute tubular necrosis, ischemia, acute interstitial nephritis, glomerulonephritis, thrombosis), pre-renal (e.g., hypovolemia, heart failure, sepsis, nephrotoxins), and postrenal causes (e.g., postrenal obstructive nephropathy, stone). AKI is independently associated with increased hospital mortality in critically ill patients, and the progression of chronic kidney disease [2-4]. Early detection and treatment of AKI can improve outcomes; however, an appropriate therapeutic strategy remains to be established.
In most cases, AKI follows transient renal ischemia and reperfusion [5]. Ischemia-reperfusion (IR) injury comes out when the blood flow to a tissue is blocked (ischemia) and then recovered (reperfusion) [6]. Typically, IR injury induces organ damage after events such as myocardial infarction, cerebrovascular occlusion and organ transplantation [7]. Despite the effort of intensive clinical and preclinical studies in renal IR injury, interventions to prevent or reduce the incidence of clinical IR injury remain elusive [8,9]. Although previous perceptions regarding this phenomenon focused on the cardiac dysfunction [10-12], a variety of ischemic disorders, including AKI, intestinal ischemia, stroke, and graft rejection after transplantation, have recently been recognized to share the common underlying pathophysiology [6].
Reactive oxygen species (ROS) and post-ischemic inflammation are considered to play a major role in the pathogenesis of IR injury, and clinical improvements could be achieved with the application of ROS-targeted therapies for AKI management [13]. It is well-established that mitochondria are the key source of ROS, generated from an explosive radical reactions upon reperfusion; these results have been observed across various tissue types, including kidney tissues [14,15]. Chouchani et al. [16] have recently identified a unifying mechanism for ROS production, during which superoxide was generated through reverse electron transport at complex I of the respiratory chain. Inflammation and oxidative stress during renal IR injury are strongly interconnected. Inflammatory cytokines are released by infiltrating leukocytes and damaged tubular cells during AKI and AKI-related oxidative stress, revealing a vicious cycle of ROS and inflammation [17,18]. The pyruvate dehydrogenase complex (PDC), catalyzes the oxidative decarboxylation of pyruvate, resulting in acetyl coenzyme A and nicotinamide adenine dinucleotide reduced in mitochondria. It is a central metabolic node of tricarboxylic acid (TCA) cycle flux and is tightly regulated in humans by any of the four pyruvate dehydrogenase kinase isoenzymes (PDK1, PDK2, PDK3, and PDK4) and pyruvate dehydrogenase phosphatase, deactivating and activating it, respectively [19,20]. PDK4 is mainly expressed in the skeletal muscle, heart, pancreatic islets, and kidneys [19, 20]. Inhibition of PDK, especially PDK4, was reported to have attenuated the development of cisplatin-induced AKI by reducing oxidative stress [21].
An in-depth understanding of the pathophysiology of AKI will facilitate the discovery and evaluation of promising therapeutic agents for application in clinical practice. In the present study, we focused on determining the role of PDK4 in renal IR injury using a diabetic mouse model. Furthermore, we assessed the effect of pharmacological PDK inhibition on ROS and inflammation following IR injury.
METHODS
Animal model of renal ischemia-reperfusion injury
All experimental procedures were performed following the appropriate institutional guidelines for animal research. The animal experiments received approval from the Institutional Animal Care and Use Committee at Kyungpook National University (IACUC reference: KNU-2020-0132).
Nine-week-old C57BL/6J male mice (DooYeol Biotech, Seoul, Korea) were administered a single intraperitoneal injection of streptozotocin (STZ, Sigma-Aldrich, St. Louis, MO, USA), 50 mg/kg daily for 5 consecutive days, to establish the diabetic mouse model. Two weeks later, STZ-induced diabetic mice with fasting blood glucose levels ≥250 mg/dL were selected. The mice were randomly divided into four groups after 20 weeks: sham controls (n=6; the kidneys were surgically exposed); STZ-induced diabetic mice (n=7); STZ-induced diabetic mice with IR injury (n=6; the kidneys were exposed and both renal pedicles were clamped for 37 minutes and reperfused for 24 hours); STZ-induced diabetic mice pretreated with dichloroacetate (DCA) treatment and subjected to IR injury (n=6). Before IR injury, DCA was administered intraperitoneally (250 mg/kg) once daily for 5 weeks.
Mice were anesthetized by an intraperitoneal injection of 50 mg/kg pentobarbital sodium (Entobar, Hanlim Pharmaceuticals, Yongin, Korea) and were placed on a heater to maintain body temperature. After performing a bilateral flank incision, the renal vein and artery were clamped for 37 minutes. For the control group, sham operations were performed without clamping of vessels. Surgical wounds were sutured and sterilized with povidone. Twenty-four hours post-surgery, the mice were sacrificed, and blood samples and both kidneys were collected. The harvested kidneys were frozen in liquid nitrogen and stored at –80°C for further analysis. Serum was obtained from blood samples using a Microtainer (BD bioscience, Franklin Lake, NJ, USA), and serum blood urea nitrogen (BUN) and serum creatinine levels were measured using Automatic Analyzer 7020 (Hitachi, Osaka, Japan).
Cell culture
NRK-52E rat kidney tubular epithelial cells were purchased from American Type Culture Collection (Manassas, VA, USA; CRL-1571) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) with high glucose (Gibco, Grand Island, NY, USA), supplemented with 5% heat-inactivated fetal bovine serum (FBS, Hyclone, Logan, UT, USA) and 1% penicillin/streptomycin (P/S, Gibco). Cells were seeded on 100 mm dishes (Corning, Kennebunk, ME, USA) at a density of 1×106 cells/plate or 60 mm dishes (Corning) at 3×105 cells/plate prior to the experiments.
Isolation of mouse primary kidney tubular cells was performed as described previously [21]. Briefly, primary mouse proximal tubules were isolated from kidneys treated with 0.1% collagenase (Gibco) dissection solution, harvested from 4-week-old male C57BL6/J (wild-type) and PDK4 knockout (KO) mice. After incubation for 2 hours, the tubules were filtered through a 200 μm nylon mesh (pluriSelect, El Cajon, CA, USA); then, cells were flushed through an 85 μm mesh (pluriSelect) using DMEM/F12 media (Gibco), 1% heat-inactivated FBS, 15 mM HEPES, 2 mM L-glutamine, 50 nM hydrocortisone (Sigma), 0.55 mM sodium pyruvate (Sigma), ITS 100X (Gibco), 10 ml/L 100× nonessential amino acid (Gibco), and P/S (Gibco). Next, tubular cells were centrifuged at 1,000 rpm for 5 minutes and resuspended in DMEM/F12. Cells were seeded on a collagen-coated plate (Corning) for 48 hours, and culture media were replaced every 2 days. On day 7, cells were split and seeded at a density of 4×105 cells in a 60 mm dish (Corning) for experimentations [21].
In vitro model of hypoxia-reoxygenation injury
In brief, NRK-52E cells were seeded on 60 mm dishes and incubated for 24 hours. Cells were then washed twice with Hanks’ Balanced Salt Solution (HBSS, Gibco) and exposed to hypoxia (1% O2) in serum-free-HBSS, with and without 2 or 5 mM DCA (Sigma), for 6 hours using a pre-conditioning hypoxia work station (Ruskinn, INVIVO2400, Pencoed, UK). After hypoxia, reperfusion was performed 2 hours after replenishing the cell cultures with normal culture media, with or without DCA (2 or 5 mM). The mouse primary tubular cells were exposed to hypoxia for 4 hours, and reperfusion was performed for 3 hours. For the knockdown experiment, hypoxia-reoxygenation (HR) was performed 16 hours after infecting the cells with adenoviral short hairpin RNA targeting green fluorescent protein (shGFP) or small hairpin PDK4 (shPDK4) (at 50 multiplicity of infection), which was amplified.
Quantitative real-time polymerase chain reaction analysis
RNA was extracted from mouse kidney tissues and NRK-52E cells using QIAzol (QIAGEN, Germantown, MD, USA), and cDNA was synthesized from 4 μg total RNA using oligo dT primer (Thermo Scientific, Vilnius, Lithuania). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the Viia7 instrument (Applied Biosystems, Foster City, CA, USA) with SYBR Green reagent (Applied Biosystems) [22]. The expression of mouse 36B4 was used as an internal control. Mouse and rat primer sequences for real-time PCR are described in Supplementary Table 1.
Western blot analysis
In brief, frozen tissues and cells were lysed with protein lysis buffer (20 mmol/L Tris-HCl [pH 7.4], 1% NP-40, 5 mmol/L ethylenediaminetetraacetic acid, 2 mmol/L Na3VO4, 100 mmol/L NaF, 10 mmol/L Na4P2O7, 100 μmol/L phenylmethylsulfonyl fluoride, 7 μg/mL aprotinin, 7 μg/mL leupeptin) and phosphatase cocktail inhibitors. Then, tissue and cell lysates were centrifuged for 10 minutes at 10,000 rpm, and supernatants were collected. The total protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Scientific). Proteins were separated on 10% to 12% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Millipore, County Cork, Ireland) [21,22]. The following antibodies were used for detecting protein expressions: anti-PDK1 (ENZO Life Science, Farmingdale, NY, USA); anti-PDK2 (Santa Cruz, Dallas, TX, USA); anti-PDK3 (AbFrontier, Seoul, Korea); anti-PDK4 (Abcam, Cambridge, MA, USA); anti-p-pyruvate dehydrogenase E1α (anti-p-PDHE1α; Ser232, Calbiochem, San Diego, CA, USA); pyruvate dehydrogenase (Thermo Scientific); anti-cleaved caspase-3 (Cell Signaling Technology, Bervely, MA, USA); β-actin (Sigma).
Histological and immunohistochemical analysis
Mouse kidneys were fixed using 4% paraformaldehyde (PFA) for 24 hours and embedded in paraffin. Then, 4 μm-thick serial sections were deparaffinized in xylene, rehydrated using descending grades of ethanol, and stained with hematoxylin & eosin (H&E), periodic acid-Schiff (PAS), neutrophil gelatinase-associated lipocalin (NGAL), and kidney injury molecule-1 (KIM-1). Immunohistochemical analysis was performed as described previously [21]. Briefly, sections were permeabilized with IHC-Tek epitope retrieval solution (IHC world, Ellicott City, MD, USA) for 45 minutes, incubated with 3% hydrogen peroxide (DUKSAN science, Seoul, Korea) for 15 minutes and blocked with UltraVision protein block (LabVision Corporation, Fremont, CA, USA) for 10 minutes. Sections were incubated for 16 hours with the primary antibody against p-PDHE1α (Ser232, Calbiochem), 4-hydroxynenenal (4-HNE, Abcam) and nitrotyrosine (NT, Millipore), NGAL (Abcam), and KIM-1 (Abcam). H&E- and PAS-stained sections were examined using light microscopy (Olympus BX53 upright microscope, Tokyo, Japan).
TUNEL staining
Paraffin-embedded 4-μm thick kidney sections were deparaffinized and stained in accordance with the manufacturer’s instructions (In situ Cell Detection Kit, Roche, Mannheim, Germany). Then, kidney sections were permeabilized with Proteinase K (20 μg/mL [Sigma-Aldrich] in 10 mM Tris/HC1, pH 7.4 to 8) for 1 hour at 37°C in an incubator. Mouse primary kidney tubular cells were fixed with 4% PFA for 15 minutes, permeabilized with 0.1% TritonX-100, and subjected to staining [21]. The kidney sections and cells were mounted using the mounting medium (Vectora Laboratories, Burlingame, CA, USA). Fluorescence images were captured using an OLYMPUS 1X81 inverted microscope system.
Annexin V analysis
NRK-52E cells were collected, resuspended in 1× binding buffer, and incubated with fluorescein isothiocyanate (FITC) and propidium iodide (PI) for 15 minutes at room temperature in the dark. Analysis was performed in accordance with the manufacturer’s instructions (FITC-Annexin V apoptosis detection kit I; BD bioscience) within 1 hour using Accuri C6 cytometer (BD bioscience).
Statistical analyses
All data are expressed as mean±standard error of the mean. Statistical analyses were performed using an unpaired Student’s t-test and P<0.05 was considered statistically significant.
RESULTS
PDK4 expression is upregulated in renal ischemia-reperfusion injury in diabetic mice
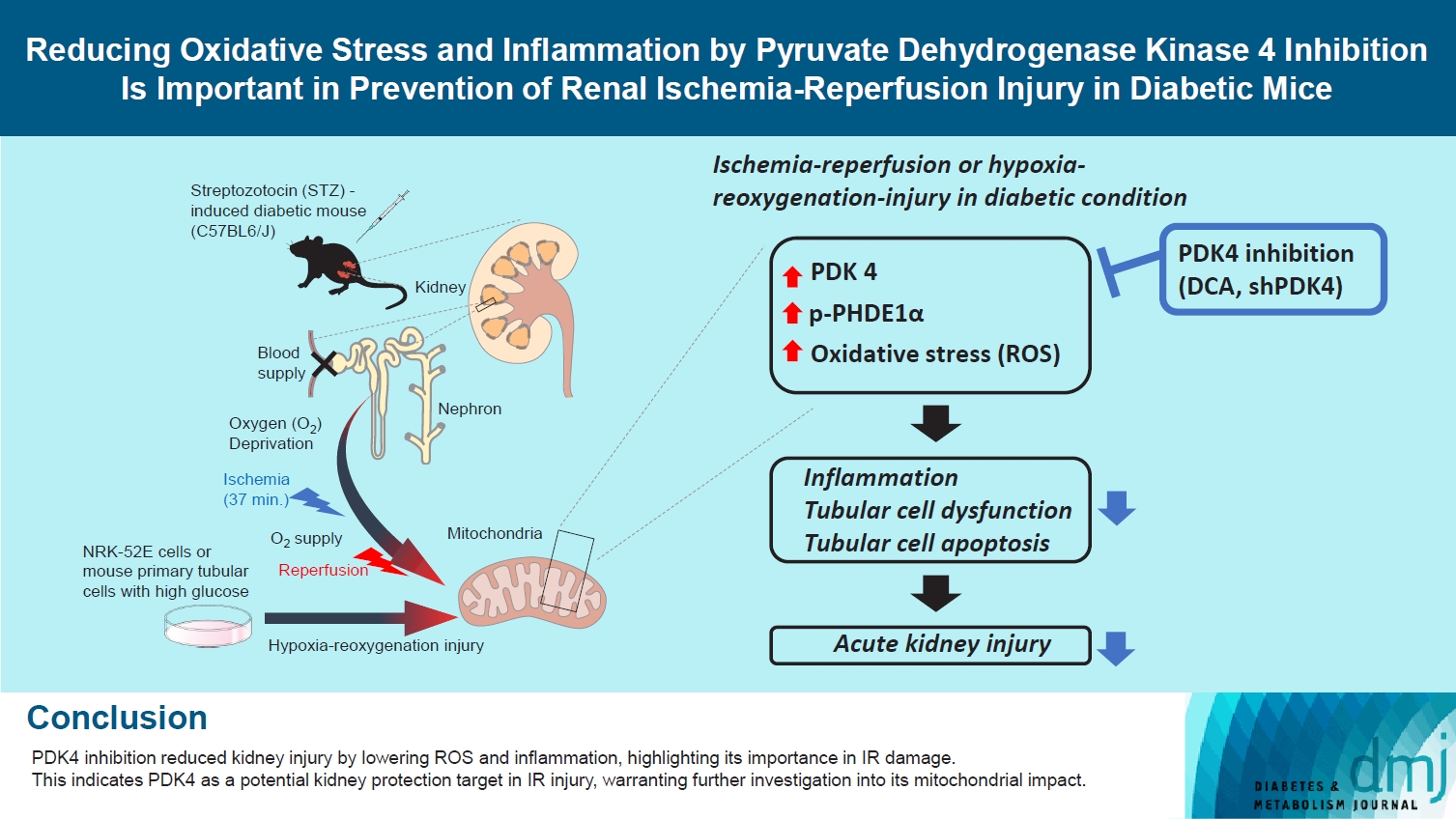
To the best of our knowledge, there are no reports indicating the role of PDK in the fundamental response of the kidney to IR; hence, we investigated the mRNA and protein levels of PDK. Herein, we employed an STZ-induced diabetic mouse model (50 mg/kg STZ administered daily for 5 days). In order to evaluate the expression of PDK during IR injury, C57BL/6 mice (9-week-old) were randomly divided into four study groups as described in previous section. Histopathological assessment of renal tissues revealed considerable tissue injury with severe tubular damage, lysis, and necrosis after IR injury (Fig. 1A). The mRNA expression of Pdk4 was significantly increased in STZ-induced diabetic mice with IR injury (Fig. 1B). Furthermore, the protein levels of PDK, as determined by Western blotting, followed the mRNA expression pattern for Pdk (Fig. 1C). The phosphorylation of PDHE1α, a target protein of PDK, was significantly increased as determined by immunohistochemical studies and Western blot analysis in STZ-induced diabetic mice with IR injury; this could be attributed to PDK4 induction (Fig. 1D and E). Accordingly, these findings demonstrate that IR injury induces the mRNA expression of Pdk4, as well as increases protein levels of PDK4 in diabetic mice.
Pyruvate dehydrogenase kinase 4 (PDK4) is induced in ischemia-reperfusion (IR) kidney injury in diabetic mice. (A) Hematoxylin and eosin (H&E) staining in mouse kidneys (original magnification ×200; scale bar, 100 μm; arrows, damaged tubules). (B) Relative mRNA level of Pdk isoforms in mice kidney tissues. (C) Protein expression and quantitative graph of PDK isoforms in mice kidney tissues. (D) Immunohistochemical image of p-pyruvate dehydrogenase E1α (p-PDHE1α) expression in mice kidney tissues (original magnification: ×200; scale bar, 100 μm; arrow, positive regions). (E) Protein expression and quantitative graph of p-PDHE1α in mice kidney tissues. Data are the mean±standard error of the mean. STZ, streptozotocin. aP<0.01 vs. Control, bP< 0.05, cP<0.01, dP<0.001 vs. STZ.
Sodium dichloroacetate mitigates IR injury
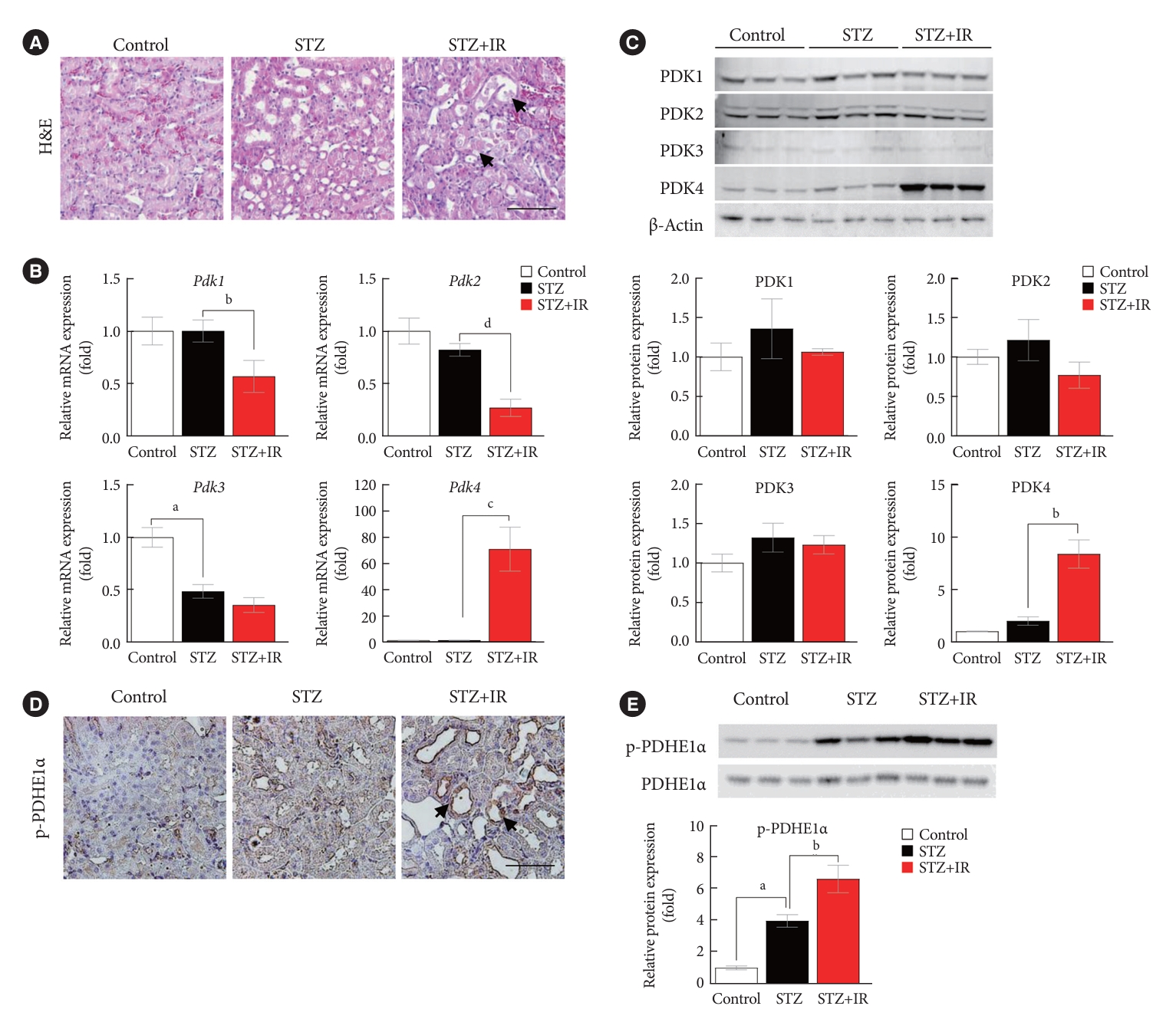
To determine whether PDK inhibition attenuates IR injury, we investigated the severity of IR renal injury and expression of PDK isoforms in diabetic mice following pretreatment with DCA, a PDK inhibitor, prior to IR injury. Our findings revealed that DCA treatment results in less severe tubular damage in diabetic mice with less extensive tubular dilatation, cellular lysis, and sloughed debris (Fig. 2A). Compared with sham-operated control mice, 37 minutes of bilateral renal ischemia followed by 24 hours of reperfusion markedly increased serum BUN and creatinine levels in STZ-induced diabetic mice (Fig. 2B). Following DCA treatment, IR injury-induced renal dysfunction was significantly attenuated in the diabetic mice. After 5 weeks of DCA treatment, no differences in blood glucose levels and body weight were observed between treated and untreated diabetic mice groups 1 day before IR injury (Supplementary Fig. 1). IR injury-induced mRNA and protein expressions of PDK4 were markedly reduced following DCA treatment (Fig. 2C and D). In line with decreased PDK4 expression, PDHE1α phosphorylation was alleviated following DCA treatment (Fig. 2E and F). These results suggest that pretreatment with DCA before IR injury mitigates renal IR injury in diabetic mice.
Sodium dichloroacetate (DCA) attenuates ischemia-reperfusion (IR) injury in diabetic mice. (A) Hematoxylin and eosin (H&E) staining, periodic acid-Schiff (PAS) staining, neutrophil gelatinase-associated lipocalin (NGAL) staining, and kidney injury molecule-1 (KIM-1) staining of mice kidney tissues (original magnification ×200; scale bar, 100 μm; arrows, damaged tubules). (B) Serum blood urea nitrogen (BUN) and creatinine in mice. (C) Relative mRNA expression of pyruvate dehydrogenase kinase (Pdk) isoforms in mice kidney tissues. (D) Protein expression of PDK isoforms in mice kidney tissues. (E) Immunohistochemical image of p-pyruvate dehydrogenase E1α (p-PDHE1α) expression in mice kidney tissues (original magnification ×200; scale bar, 100 μm; arrow, positive regions). (F) Protein expression of p-PDHE1α in mice kidney. Data are the mean±standard error of the mean. STZ, streptozotocin. aP<0.01 vs. Control, bP<0.01 vs. STZ+IR, cP<0.01 vs. STZ, dP<0.05 vs. STZ+IR, eP<0.05 vs. STZ.
DCA and PDK knockdown attenuates IR-induced apoptosis of renal cells in diabetic mice
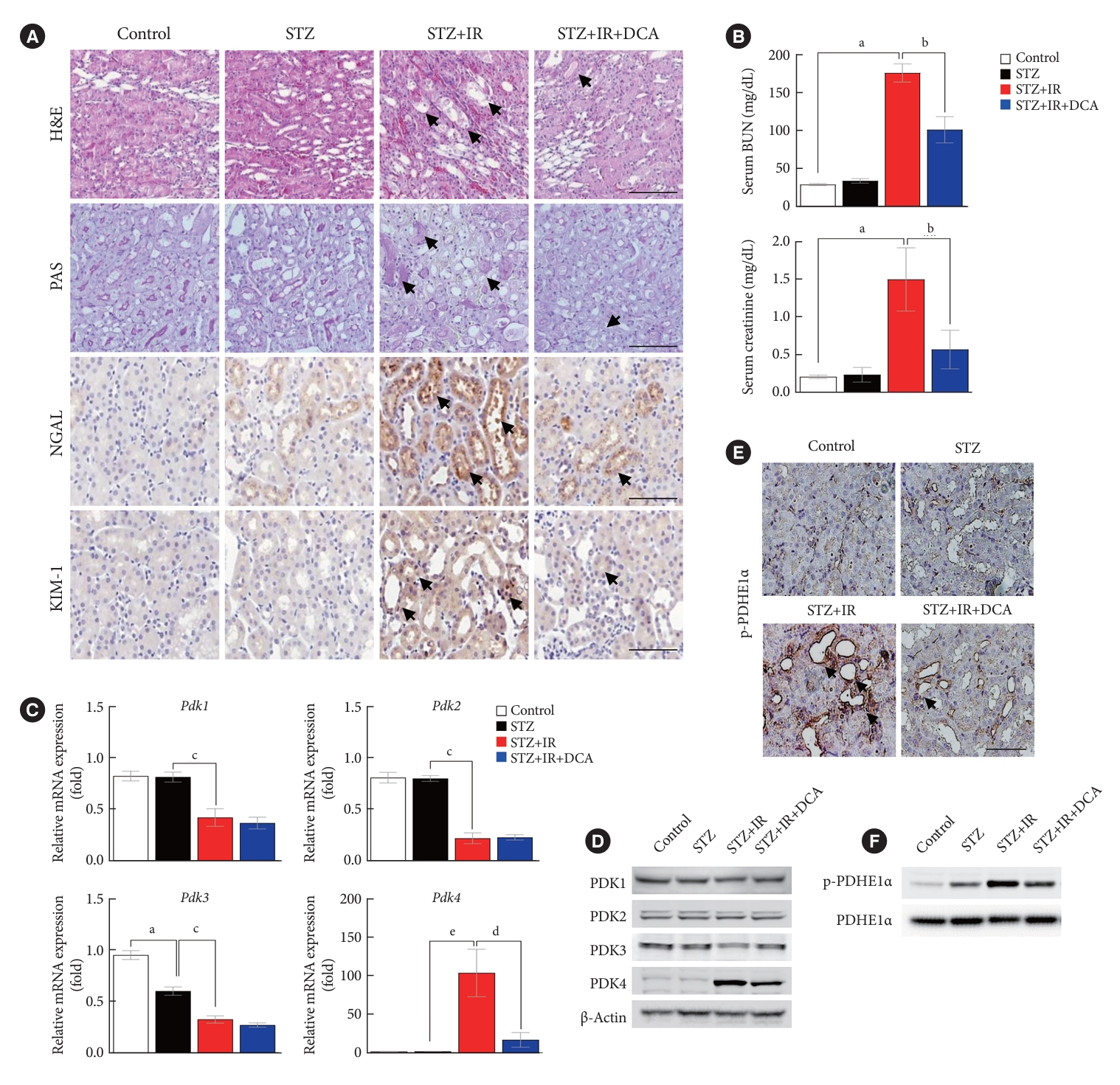
Renal IR injury is associated with cellular apoptosis, which might contribute to renal dysfunction. To evaluate the effect of PDK inhibition on the apoptosis of renal cells in an IR renal injury model using diabetic mice, we used the TdT-mediated dUTP nick end labeling (TUNEL) assay and flow cytometry analysis with Annexin V/PI staining in mouse primary tubular cells and NRK-52E cells. In addition, to determine the mechanism through which PDK contributes to IR injury of diabetic kidney tissues, we established an in vitro model using high glucose-conditioned NRK-52E and mouse primary tubular cells, which were incubated using the hypoxia work station for 6 hours followed by reoxygenation for 2 hours. NRK-52E cells with HR revealed significant apoptotic induction (Supplementary Fig. 2A). Following HR, Pdk4 mRNA expression was increased in both NRK-52E cells and mouse primary tubular cells (Supplementary Fig. 2B and C). Compared with mice without IR injury, apoptotic cells, quantified using the TUNEL assay, were increased in STZ-induced diabetic mice with IR injury (Fig. 3A). The protein level of cleaved caspase-3 was markedly increased in diabetic mice with IR injury when compared with that in diabetic mice without IR injury; DCA treatment decreased this level (Fig. 3B). The reduced number of apoptotic cells and decreased cleaved caspase-3 protein levels following DCA treatment, indicate the potential reno-protective effect of PDK inhibition after IR injury. Additionally, Annexin V/PI staining revealed that HR injury could strongly induce apoptosis and necrosis of NRK-52E cells; DCA treatment markedly reduced the degree of cell death (Fig. 3C). Based on TUNEL assay results for mouse primary tubular cells, the presence of apoptotic cells was lower in DCA-treated cells than in DCA untreated cells (Fig. 3D). In both NRK-52E and mouse primary tubular cells, DCA treatment markedly decreased protein levels of cleaved caspase-3 in a dose-dependent manner when compared with those of untreated cells (Fig. 3E and F). Furthermore, the protein level of cleaved caspase-3 was considerably reduced following shPDK4 treatment of NRK-52E cells upon HR (Fig. 3G). To further verify the role of PDK4 in hypoxic kidney damage, cleaved caspase-3 levels were analyzed in primary tubular cells isolated from PDK4 KO mice. Protein levels of cleaved caspase-3 were reduced in primary tubular cells derived from PDK4 KO mice when compared with those in wild-type mice (Fig. 3H).
Sodium dichloroacetate (DCA) and pyruvate dehydrogenase kinase (PDK) knockdown attenuates ischemia-reperfusion (IR)-induced apoptosis in mice and NRK-52E cells. (A) TdT-mediated dUTP nick end labeling (TUNEL) staining in mice kidney tissues (original magnification ×200; scale bar, 200 μm; arrows, TUNEL positive tissues). (B) Protein expression and quantitative graph of cleaved caspase-3 in mice. (C) Representative Annexin V/propidium iodide (PI) staining in NRK-52E cells. (D) TUNEL staining in mouse primary tubular cells (original magnification ×200; scale bar, 100 μm; arrows, TUNEL positive cells). Protein expression of cleaved caspase-3 in NRK-52E cells (E) and mouse primary tubule cells (F). (G) Protein expression of cleaved caspase-3 in NRK-52E cells infected with adenoviral short hairpin RNA targeting green fluorescent protein (shGFP) or small hairpin PDK4 (shPDK4). (H) Protein expression of cleaved caspase-3 in mouse primary tubular cells isolated from wild-type (WT) and PDK4 knockout (KO) mice. Data are the mean±standard error of the mean. STZ, streptozotocin; PE, phycoerythrin; FITC, fluorescein isothiocyanate; HR, hypoxia-reoxygenation. aP<0.01 vs. Control, bP<0.01 vs. STZ, cP<0.01 vs. STZ+IR, dP<0.01 vs. HR.
DCA attenuates IR-induced oxidative stress and inflammation in diabetic mice and NRK-52E cells
As ROS and inflammation are recognized as essential effectors of IR injury, we investigated the production of ROS and inflammatory mediators in a murine model of IR injury with diabetes [23,24]. Fig. 4A and B present the expression of oxidative stress markers, 4-HNE and NT in the kidneys of sham control mice, STZ-induced diabetic mice, STZ-induced diabetic mice with IR injury, and STZ-induced diabetic mice pretreated with DCA, followed by IR injury. We observed that the STZ-induced diabetic mice with IR injury exhibited a marked increase in 4-HNE and NT staining. Furthermore, DCA treatment for 5 weeks prior to IR injury significantly decreased the 4-HNE and NT burden.
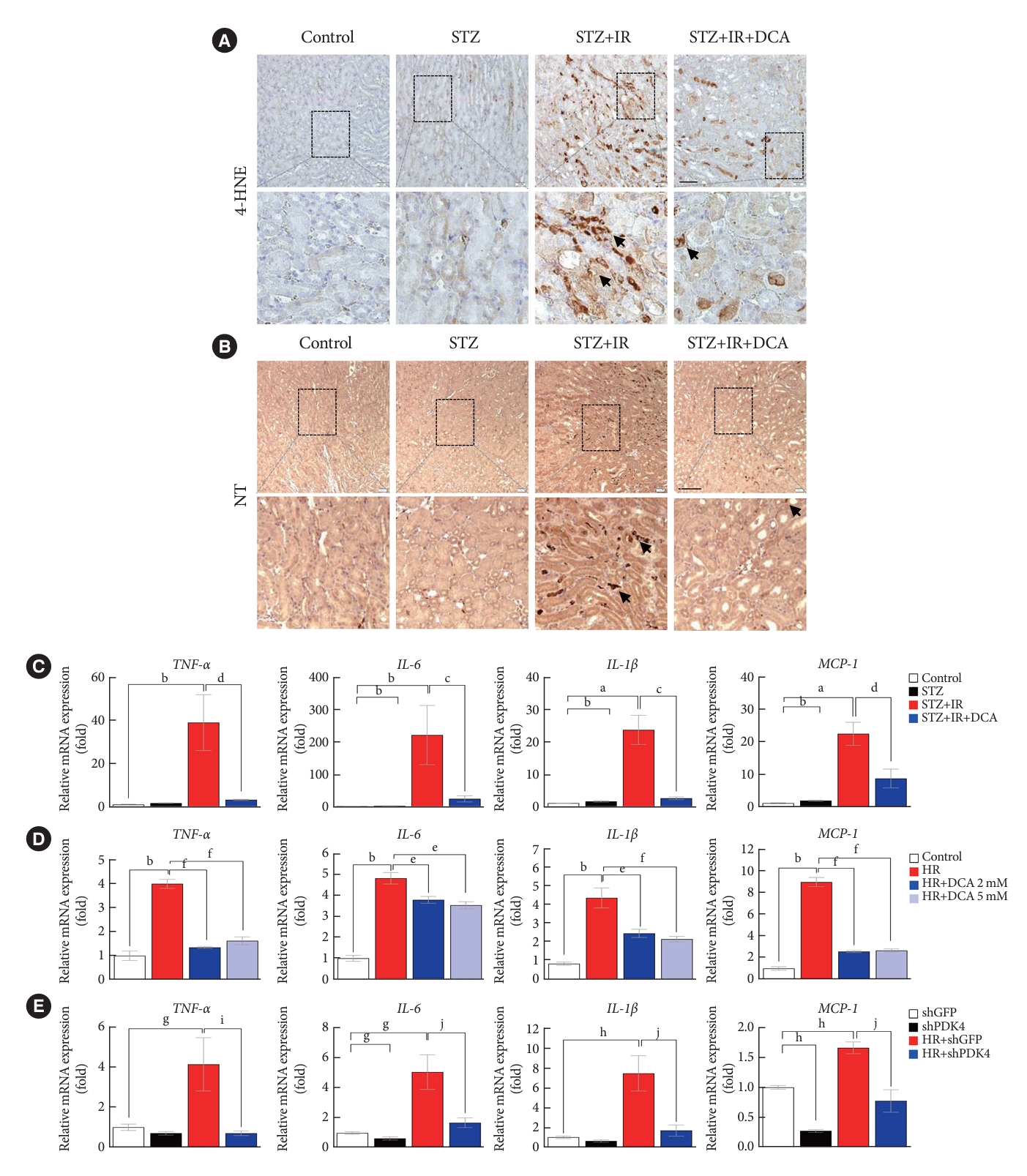
Sodium dichloroacetate (DCA) attenuates ischemia-reperfusion (IR)-induced oxidative stress and inflammation in diabetic mice and NRK-52E cells. (A) 4-Hydroxynenenal (4-HNE) staining in mice kidney tissues (original magnification ×200; scale bar, 100 μm arrows, 4-HNE positive areas). (B) Nitrotyrosine (NT) staining in mice kidney tissues (original magnification ×100; scale bar, 50 μm arrows, NT positive areas). (C) Inflammatory markers in mice. (D) Inflammatory markers in NRK-52E with or without DCA. (E) Inflammatory markers in NRK-52E cells infected with adenoviral short hairpin RNA targeting green fluorescent protein (shGFP) or small hairpin pyruvate dehydrogenase kinase 4 (shPDK4). Data are the mean±standard error of the mean. STZ, streptozotocin; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; IL-1β, interleukin-1β; MCP-1, monocyte chemoattractant protein-1; HR, hypoxia-reoxygenation. aP<0.05, bP<0.01 vs. Control, cP<0.05, dP<0.01 vs. STZ+IR, eP<0.05, fP<0.01 vs. HR, gP<0.05, hP<0.01 vs. shGFP, iP<0.05, jP<0.01 vs. HR+shGFP.
Assessment of inflammatory cytokines revealed that STZ-induced diabetic mice with IR injury exhibited higher expression levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β), and monocyte chemoattractant protein-1 (MCP-1) when compared with those without IR injury; DCA treatment alleviated this effect (Fig. 4C). In NRK-52E cells, DCA or shPDK4 treatment markedly reduced the expression levels of TNF-α, IL-6, IL-1β, and MCP-1 when compared with those observed in untreated cells (Fig. 4D and E). These findings suggest that PDK4 blockade possesses a potent reno-protective effect by mitigating the renal oxidative stress and extensive inflammation observed post-IR injury.
DISCUSSION
A burst of ROS and post-ischemic inflammation is recognized as fundamental to IR injury, as it generates downstream tissue injury. It is well-established that most cellular ROS are produced from mitochondria and glucose metabolism. PDKs phosphorylate and inactivate PDC, an inner-mitochondrial-membrane enzyme complex that regulates the entry point of pyruvate into the TCA cycle. PDKs reportedly demonstrate tissue-specific abundance and are distinctly regulated manner in mammals. PDK1 is responsible for the Warburg effect in cancer cells, which is associated with cancer metabolism. It has been reported as one of the target genes of hypoxia-inducible factor-1 (HIF-1), which is an oxygen-sensing transcription factor, under hypoxic conditions [25,26]. Activation of glycolytic genes by HIF-1 is considered pivotal for cellular adaptation to low oxygen concentrations via the increased conversion of glucose to lactate. Upregulation of PDK3 by HIF-1 has been observed in cancer cell lines under hypoxic conditions as well as in proliferative stem cells for metabolic adaptation to build biomass and preserve the redox balance [27-29]. Previous studies have demonstrated the upregulation of PDK4 in several peripheral tissues, including the heart, skeletal muscle, adipose tissues, and kidneys in starved states [30-32]. PDK4 protein levels are relatively low in the tissues of a well-fed mouse. However, these levels were elevated under conditions such as fasting, when down-regulation of aerobic glucose oxidation is required to spare pyruvate for gluconeogenesis [33,34]. Furthermore, PDK4 is dramatically increased in the liver, heart, and skeletal muscle under diabetic conditions, in high inorganic phosphate-treated vascular smooth muscle cells, and cisplatin-injured renal tissues under extremely stressed conditions [21,22,30,35-37]. Oxidative stress, which predominantly results from excessive ROS accumulation, plays an essential role in the pathogenesis of cisplatin-induced AKI [21]. The present study revealed that PDK4 is also upregulated after IR-induced renal injury and tubular damage that causes cellular apoptosis. The expression of other PDK isoforms showed no significant change following IR injury. Furthermore, kidney injury and tubular apoptosis were significantly attenuated by reduced ROS production and inflammation upon PDK inhibition, associated with a marked decrease in PDK4 expression. Our findings indicate that PDK4 is mechanistically linked with cellular ROS production in renal tissue via stressful stimuli, such as IR injury or cisplatin. Although the underlying causes of AKI vary widely, PDK4 modulation seems to play a critical role in AKI attenuation.
AKI is common in diabetes and potentially causes chronic kidney disease or end-stage renal disease. Recent large cohort studies have reported that AKI may result in severe consequences in patients with diabetes; resulting in significantly lower recovery rates, higher rates of in-hospital mortality, and progression to advanced chronic kidney disease, independent of other risk factors [38,39]. Using animal models, Goor et al. [40] and Peng et al. [41] have previously reported the high vulnerability of STZ-induced diabetic rats or mice to IR renal injury. Ongoing studies are investigating mechanisms underlying accelerated kidney injury in diabetes mellitus with AKI. A preclinical study evaluating the long-term effects of acute renal ischemic injury in obese diabetic rats has revealed that postischemic inflammation is a crucial factor in the acceleration of chronic kidney disease in obesity/diabetes [42]. Moreover, Gao et al. [43] have documented that the ischemic AKI susceptibility of diabetic mice can be repressed by a TNF-α neutralizing antibody, thereby providing evidence supporting the role of the inflammatory response mediated by TNF-α. In the present study, we revealed that ischemia and reperfusion enhance PDK4 expression in the kidney of a diabetic mouse model, as well as increase oxidative stress and inflammation induced kidney injury. The mRNA expression of TNF-α markedly increased after renal ischemia and reperfusion; this indicates that TNF-α has a possible role as a mediator enhancing the susceptibility of diabetic mice to IR injury. A PDK inhibitor profoundly alleviated the IR injury-induced expression of TNF-α. In addition, IR injury increased the mRNA expression of MCP-1, which has been associated with the development of tissue fibrosis. MCP-1 is a chemokine that activates monocytes and macrophages and mediates tubulointerstitial inflammation that precedes fibrosis [44,45]. MCP-1 stimulates the expression of IL-6, cell adhesion molecules, and other inflammatory factors that contribute to renal tubular fibrosis [46]. Herein, PDK4 inhibition also attenuated the increased expression of MCP-1 following IR injury. These findings indicate that the expression of TNF-α, IL-6, IL-1β, and MCP-1 increases with enhanced PDK4 expression following IR injury. Furthermore, the decreased expression of inflammatory or fibrosis markers after PDK inhibition suggests the fact that PDK4 plays a critical role in HR damage in diabetic mice.
Although this study presented crucial evidence on the role of PDK4 in IR injury, it also has certain limitations. First, we did not compare the severity of IR injury between control mice and STZ-induced diabetic mice with IR injury. As the vulnerability of animals or patients with diabetes to AKI caused by various factors has been well documented, we specifically examine the role of PDK4 in renal IR injury of diabetic mice. Second, more specific and detailed signaling pathways, as well as related mediators associated with high PDK4 expression in kidney IR injury, were not evaluated in the present study. Detailed molecular studies are needed to delineate the upstream mechanism of increased PDK4 expression in kidney IR injury. Third, the PDK4 inhibitor was administered prior to IR injury; accordingly, the impact of PDK4 inhibitor treatment post-IR remains unknown. Pretreatment with the PDK4 inhibitor may be beneficial in AKI-prone conditions. Finally, due to species differences between mice and humans, this result needs to be cautiously interpreted. Although many issues remained unsolved, our findings still provide a potential strategy for preventing AKI.
In summary, our findings indicate that IR injury enhances PDK4 expression in the kidney of the diabetic mouse model. PDK4 inhibition mitigates kidney injury by decreasing ROS production and inflammation, suggesting a critical role for PDK4 in hypoxia-oxygenation damage. Collectively, these results suggest that inhibition of PDK4 is an imperative target for reno-protection during IR injury. The role of PDK4 needs to be further elucidated and established in terms of mitochondrial function and dynamics in renal IR injury.
SUPPLEMENTARY MATERIALS
Supplementary materials related to this article can be found online at https://doi.org/10.4093/dmj.2023.0196.
Sequences of primers used for quantitative real-time polymerase chain reaction
Blood glucose levels and body weight before ischemia-reperfusion (IR) injury are unaltered by long-term dichloroacetate (DCA) treatment in streptozotocin (STZ)-induced diabetic mice. Blood glucose level (A) and body weight (B) were measured on 1 day before IR injury. Data are the mean±standard error of the mean. aP<0.01 vs. Control.
Pyruvate dehydrogenase kinase 4 (PDK4) is increased in NRK-52E and mouse primary tubular cells after hypoxia-reoxygenation (HR). (A) Cell morphology of NRK-52E (original magnification ×40; arrows, apoptotic cells). (B) Relative mRNA expression of Pdk isoforms in NRK-52E (hypoxia, 6 hours; reoxygenation, 2 hours). (C) Relative mRNA expression of Pdk isoforms in mouse primary tubular cells (hypoxia, 4 hours; reoxygenation, 3 hours). Data are the mean±standard error of the mean. LG, low glucose; HG, high glucose; HR, hypoxia-reoxygenation. aP<0.01 vs. LG, bP<0.01 vs. HG.
Notes
CONFLICTS OF INTEREST
Sung Hee Choi has been associate editor of the Diabetes & Metabolism Journal since 2022. She was not involved in the review process of this article. Otherwise, there was no conflict of interest.
AUTHOR CONTRIBUTIONS
Conception or design: D.H.K., C.J.O., I.K.L.
Acquisition, analysis, or interpretation of data: A.R.K., D.H.K., M.J.K., C.J.O.
Drafting the work or revising: A.R.K., M.J.K., C.J.O., J.H.J.
Final approval of the manuscript: A.R.K., D.H.K., M.J.K., C. J.O., J.H.J., S.H.C., I.K.L.
FUNDING
This research was supported by the Korea Health Technology R&D Project through KHIDI, backed by the Ministry of Health & Welfare (HI16C1501 and HR22C1832), the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2022R1A2B5B03001929, NRF-2021R1 A5A2021614 and NRF-2022R1C1C1010898), the Korea Drug Development Fund sponsored by Ministry of Science and ICT, Ministry of Trade, Industry, and Energy, and Ministry of Health and Welfare (HN21C0923000021), and a grant from the Korean Diabetes Association (Min-Ji Kim, 2021F-3).
Acknowledgements
None