Dyslipidemia in Patients with Chronic Kidney Disease: An Updated Overview
Article information

Abstract
Dyslipidemia is a potentially modifiable cardiovascular risk factor. Whereas the recommendations for the treatment target of dyslipidemia in the general population are being more and more rigorous, the 2013 Kidney Disease: Improving Global Outcomes clinical practice guideline for lipid management in chronic kidney disease (CKD) presented a relatively conservative approach with respect to the indication of lipid lowering therapy and therapeutic monitoring among the patients with CKD. This may be largely attributed to the lack of high-quality evidence derived from CKD population, among whom the overall feature of dyslipidemia is considerably distinctive to that of general population. In this review article, we cover the characteristic features of dyslipidemia and impact of dyslipidemia on cardiovascular outcomes in patients with CKD. We also review the current evidence on lipid lowering therapy to modify the risk of cardiovascular events in this population. We finally discuss the association between dyslipidemia and CKD progression and the potential strategy to delay the progression of CKD in relation to lipid lowering therapy.
INTRODUCTION
Dyslipidemia is a potentially modifiable cardiovascular risk factor. As high low-density lipoprotein cholesterol (LDL-C) levels [1-4] as well as high triglycerides (TG) [5] and low high-density lipoprotein cholesterol (HDL-C) [6,7] levels are significantly associated with the risk of cardiovascular events in the general population, trials have aimed to correct lipid profiles in patients with dyslipidemia for the primary and secondary preventions of cardiovascular events. On the basis of currently available data, most clinical guidelines on dyslipidemia primarily target to lower LDL-C levels [8-10].
Whereas the recommendations for the treatment target of dyslipidemia in the general population are being more and more rigorous, the 2013 Kidney Disease: Improving Global Outcomes (KDIGO) clinical practice guideline for lipid management in chronic kidney disease (CKD) presented a relatively conservative approach with respect to the indication of lipid lowering therapy and therapeutic monitoring among the patients with CKD [11]. This may be largely attributed to the lack of high-quality evidence derived from CKD population, among whom the overall feature of dyslipidemia is considerably distinctive to that of general population. On top of the characteristic alterations in the lipid profile among the patients with CKD [2-4,12], the impact of dyslipidemia on cardiovascular outcomes seems to be substantially modified by deteriorating kidney function [13,14]. Most importantly, accumulating evidence strongly suggests the association between dyslipidemia and the risk of CKD progression [15-17].
This review article covers the characteristic features of dyslipidemia and the impact of dyslipidemia on cardiovascular outcomes in patients with CKD. We also review the current evidence on lipid lowering therapy to modify the risk of cardiovascular events in this population. We finally discuss the association between dyslipidemia and CKD progression and the potential strategy to delay the progression of CKD in relation to lipid lowering therapy.
FEATURES OF DYSLIPIDEMIA IN CKD
Dyslipidemia is a common complication of CKD [16,18,19], although the prevalence of dyslipidemia in Korean patients with CKD has not been precisely surveyed yet. As the Korean Society of Lipid and Atherosclerosis reported that, in 2018, the prevalence of dyslipidemia was 45.6% in men and 31.3% in women among the general population [20], the prevalence in CKD population is supposed to be higher than that. Uremic condition leads to various alterations in the quantity and quality of circulating lipoproteins. Typically, dyslipidemia in patients with CKD is featured with low HDL-C levels and high TG levels, as well as the other compositional changes in the lipoproteins [2-4,12]. In the overall, such changes are believed to promote atherosclerosis, contributing to high cardiovascular burden in patients with CKD [2-4].
Structural changes in LDL-C
Cholesterol derived from apolipoprotein B (ApoB), such as low-density lipoprotein (LDL), accumulates in the subintimal space of vascular beds, and is critical to initiate the subsequent atherosclerotic processes (Fig. 1) [21,22]. Therefore, lowering LDL-C levels has been a major strategy to reduce cardiovascular risk in the general population as well as in patients with CKD [1-4]. It is important to note that; however, LDL and total cholesterol levels are not dramatically increased among the patients with CKD [2,3,23,24]. Rather, a structural change is remarkable in LDL-C, with predominance of small dense LDL particles [25]. Due to their increased capacity to penetrate the arterial intima and increased susceptibility to oxidation, small dense LDL particles are known to be more atherogenic than the other LDL subfractions [22,26,27].
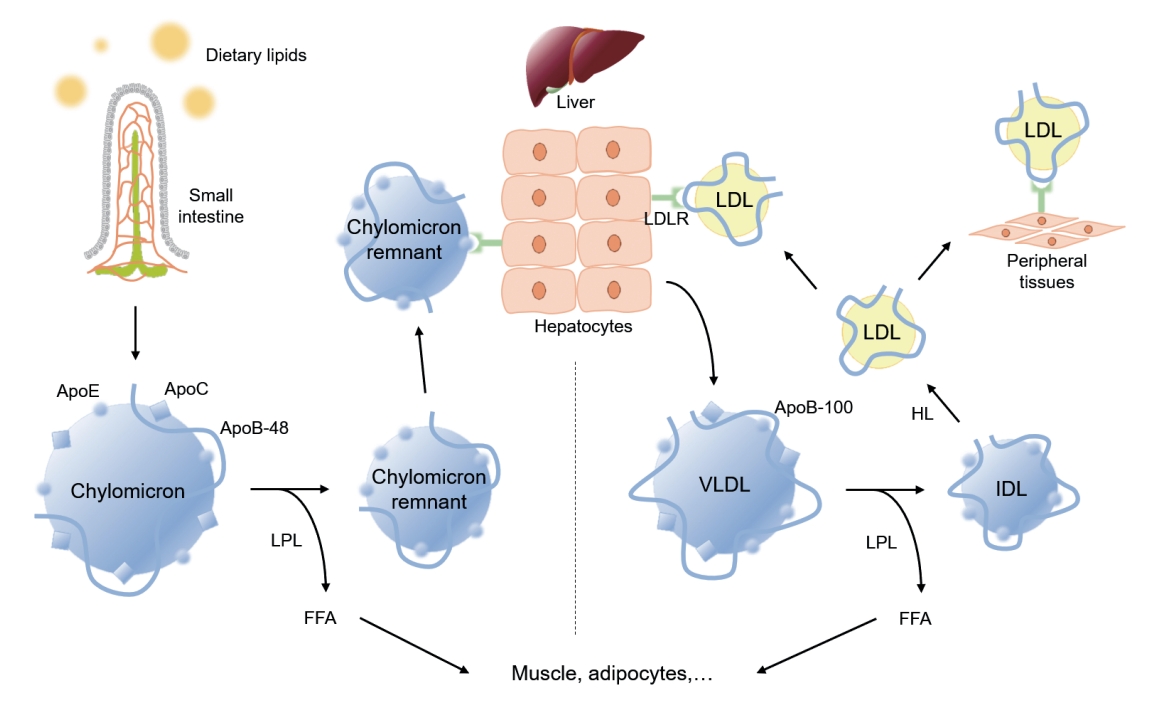
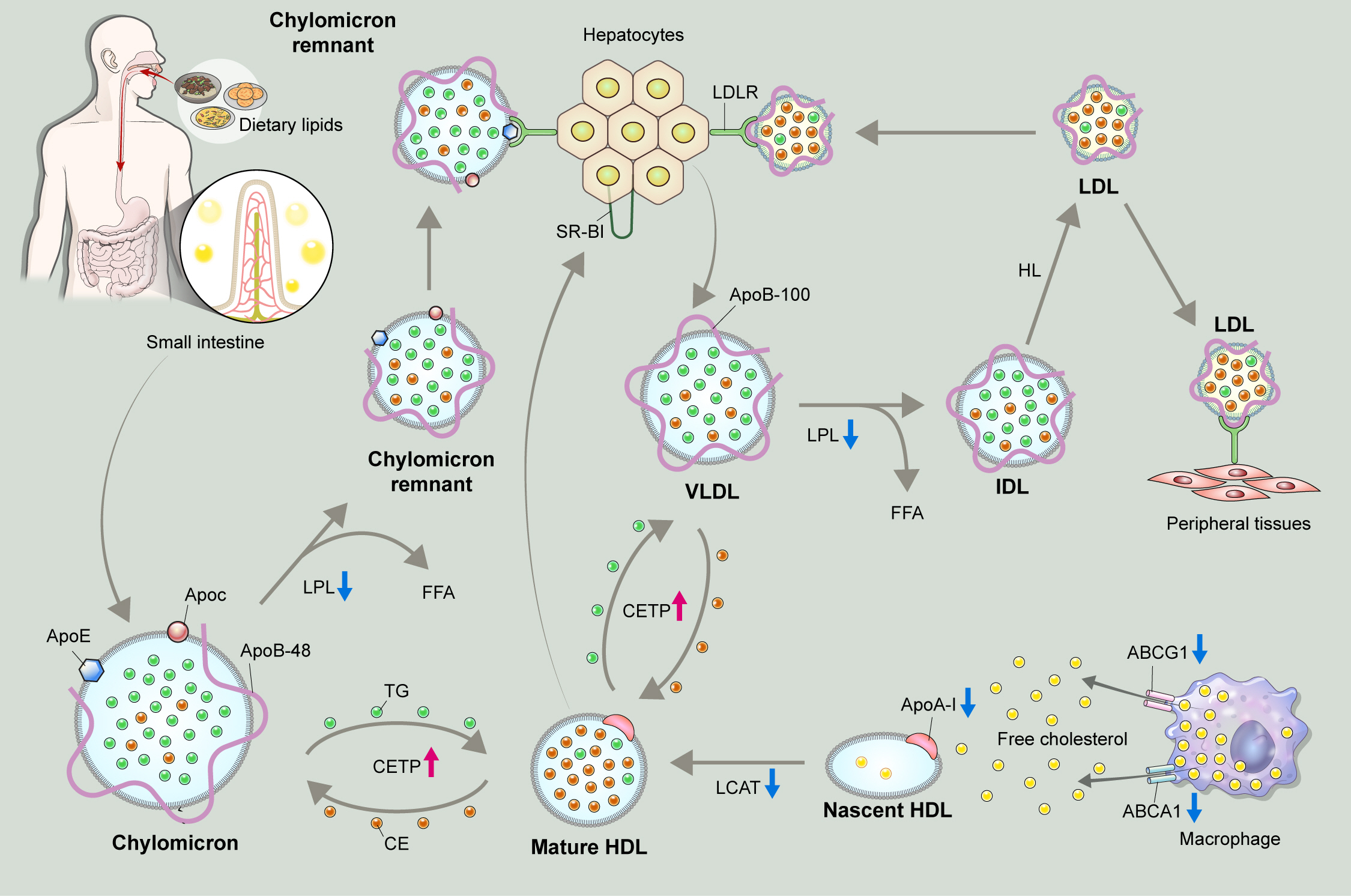
Metabolic pathway of exogenous and endogenous lipoproteins. The exogenous pathway involves trafficking of dietary lipids to peripheral organs, and ultimately to the liver. Long chain fatty acids derived from hydrolyzed dietary lipids are transformed to triglyceride (TG) in lymphatic endothelial cells, and are packaged with, apolipoprotein B-48 (ApoB-48) as well as apolipoprotein E (ApoE) and apolipoprotein C (ApoC) to form chylomicrons. Chylomicrons secreted into intestinal lymphatic circulation reach systemic circulation via thoracic duct. Chylomicrons are hydrolyzed by lipoprotein lipase (LPL) that is anchored on the surface of capillary endothelium in adipose tissue, and heart and skeletal muscles to release free fatty acids (FFAs), and shrink to chylomicron remnants. Chylomicron remnants bind to low-density lipoprotein receptor (LDLR) thorough ApoE-mediated mechanism, and are rapidly cleared from the circulation. The endogenous pathway delivers hepatically derived lipoproteins to the periphery, providing energy source during fasting. Hepatocytes esterify fatty acids to form TG, which are packaged into very low-density lipoprotein (VLDL) particles with apoB-100. Circulating VLDL particles are hydrolyzed by LPL to release FFAs, and are referred to as intermediate-density lipoprotein (IDL). About 50% of IDL particles are removed by hepatocytes via ApoE-mediated binding to LDLR. The remainder of IDL particles are further processed by hepatic lipase (HL) to form low-density lipoprotein (LDL) particles. LDL particles may be removed from the circulation by hepatocytes via apoB-100-mediated binding to LDLR, or may serve as a source of cholesterol deposit in the peripheral tissue, triggering a subsequent atherosclerotic process.
Defective metabolism in HDL-C
HDL-C is involved in the process of reverse cholesterol transport to clear excess cholesterol particles from peripheral tissue, and has been known to be protective against atherosclerotic processes (Fig. 2) [23,28]. The synthesis and maturation of high-density lipoprotein (HDL) is deranged in multiple aspect under uremic conditions. First, the hepatic synthesis of apolipoprotein A-I (ApoA-I), a major apolipoprotein of HDL is significantly reduced [23,29]. Second, the efflux of free cholesterol particles from tissue macrophages via ATP-binding cassette (ABC) transporters, such as ABCA1 and ABCR1, is interrupted due to the downregulation of the transporters, resulting in the defective maturation of nascent HDL [23,30]. Third, both plasma levels and activity of lecithin-cholesterol acyltransferase (LCAT), which converts free cholesterol to cholesteryl ester (CE) to form mature HDL-C, is reduced [31,32]. Furthermore, the activity of cholesteryl ester transfer protein (CETP) is enhanced in patients with CKD [23,33]. As, CETP medicates exchange of TG and CE between HDL and ApoB lipoproteins, enhanced CETP activity leads to reduction in the HDL-C levels. As a net effect, the plasma levels and the maturation of HDL-C are significantly impaired in patients with CKD, contributing to the suboptimal anti-oxidative and anti-inflammatory function of HDL particles under uremic condition [34,35].
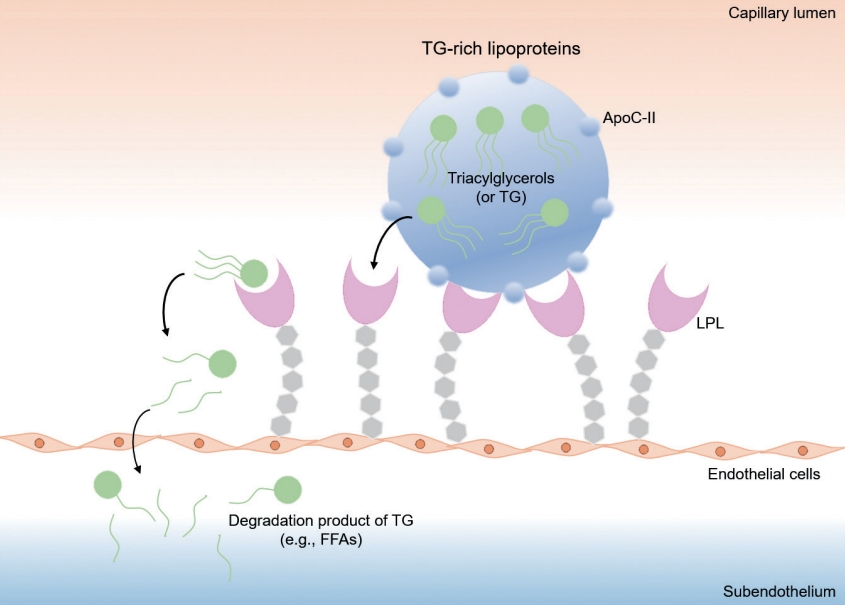
Schematic diagram for the function of lipoprotein lipase (LPL). LPL is anchored on the surface of capillary endothelial cells, and plays a key role in the hydrolysis of triglyceride (TG)-rich lipoproteins (i.e., chylomicron and very low-density lipoprotein [VLDL]). Apolipoprotein C-II (ApoC-II) on the surface of TG-rich lipoproteins is a co-factor required to activate LPL. LPL degrades TG to release two free fatty acids (FFAs) and one monoacylglycerol molecule, and transforms chylomicron and VLDL into chylomicron remnants and intermediate-density lipoprotein.
Delayed catabolism of TG-rich lipoproteins
As TG is a main component of TG-rich lipoproteins, such as very low-density lipoprotein (VLDL) and chylomicrons (CM), its plasma level reflects circulating quantity of TG-rich lipoproteins [36,37]. Circulating TG-rich lipoproteins bind to endothelial cells in vascular beds, triggering local inflammation characterized by upregulation of integrins, generation of reactive oxygen species, cytokine production, and complement activation [38,39]. Hypertriglyceridemia is an early finding of CKD, and is one of the most common features of dyslipidemia in patients with CKD [40]. Elevation in plasma TG levels is primarily attributed to the delayed catabolism of TG-rich lipoproteins. TG packed in VLDL or CM is hydrolyzed to free fatty acid by lipoprotein lipase (LPL) (Fig. 3) [41]. The expression of LPL is downregulated under uremic condition [42]. Moreover, the activity of LPL is also reduced in patients with CKD, because of parathyroid hormone-induced insulin resistance [43] and upregulation of its competitive inhibitor, such as apolipoprotein C-III (ApoC-III) [44]. Clearance of circulating VLDL particles is also defective under uremia due to the downregulation of VLDL receptors in adipocytes and myocytes [45]. Defective metabolism of major lipoproteins in CKD is summarized in Fig. 4.
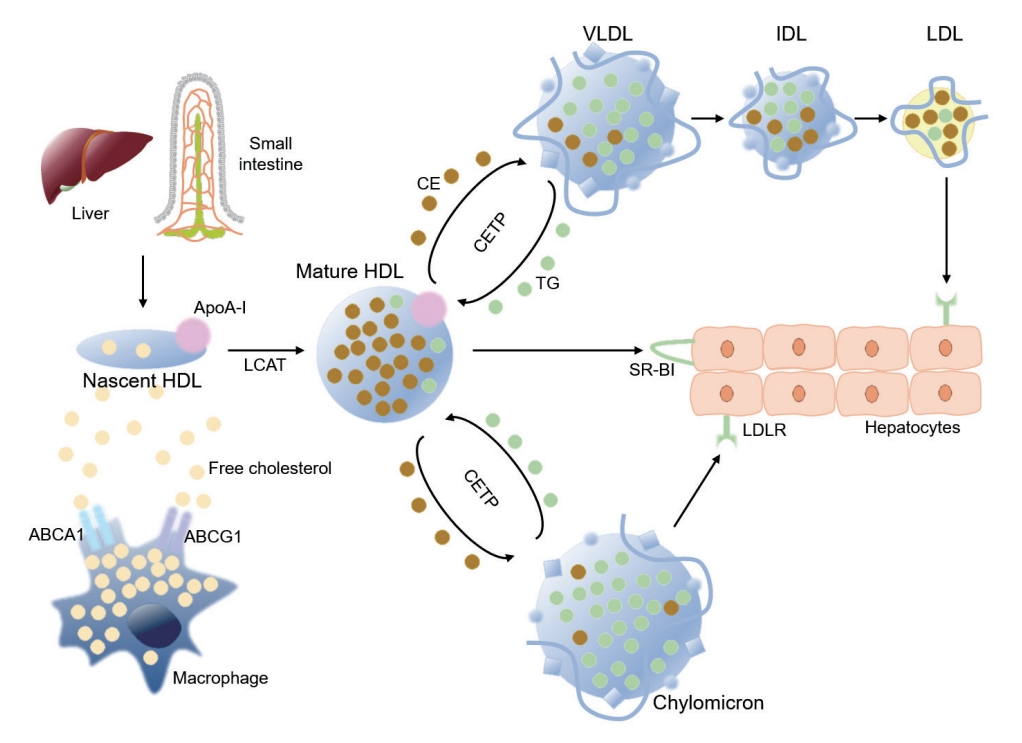
High-density lipoprotein (HDL) metabolism in reverse cholesterol transport. Apolipoprotein A-I (ApoA-I) is secreted by liver and intestine, as nascent HDL particles. Free cholesterol is acquired from macrophages and other peripheral cells, where ApoA-I promotes the efflux of free cholesterol from macrophages via ATP-binding cassette subfamily member 1 (ABCA1). The efflux of free cholesterol from macrophages is also mediated by ATP-binding cassette subfamily G member 1 (ABCG1). Lecithin-cholesterol acyltransferase (LCAT), a plasma enzyme associated with HDL, esterifies free cholesterol to cholesteryl ester (CE) inside HDL particle to form mature HDL particles. CE inside mature HDL particles can be untaken by hepatocytes via scavenger receptor class B type I (SR-BI), a receptor for HDL that mediates the selective transfer of CE to hepatocytes and recycling of the dissociated HDL particles. Alternatively, CE inside mature HDL can be transferred to apolipoprotein B-containing lipoproteins, such as very low-density lipoprotein (VLDL) and chylomicron that are rich with triglyceride (TG), in exchange of TG by cholesteryl ester transfer protein (CETP), and then cleared the circulation by low-density lipoprotein receptor (LDLR)-mediated endocytosis of hepatocytes. LDL, low-density lipoprotein; IDL, intermediate-density lipoprotein .
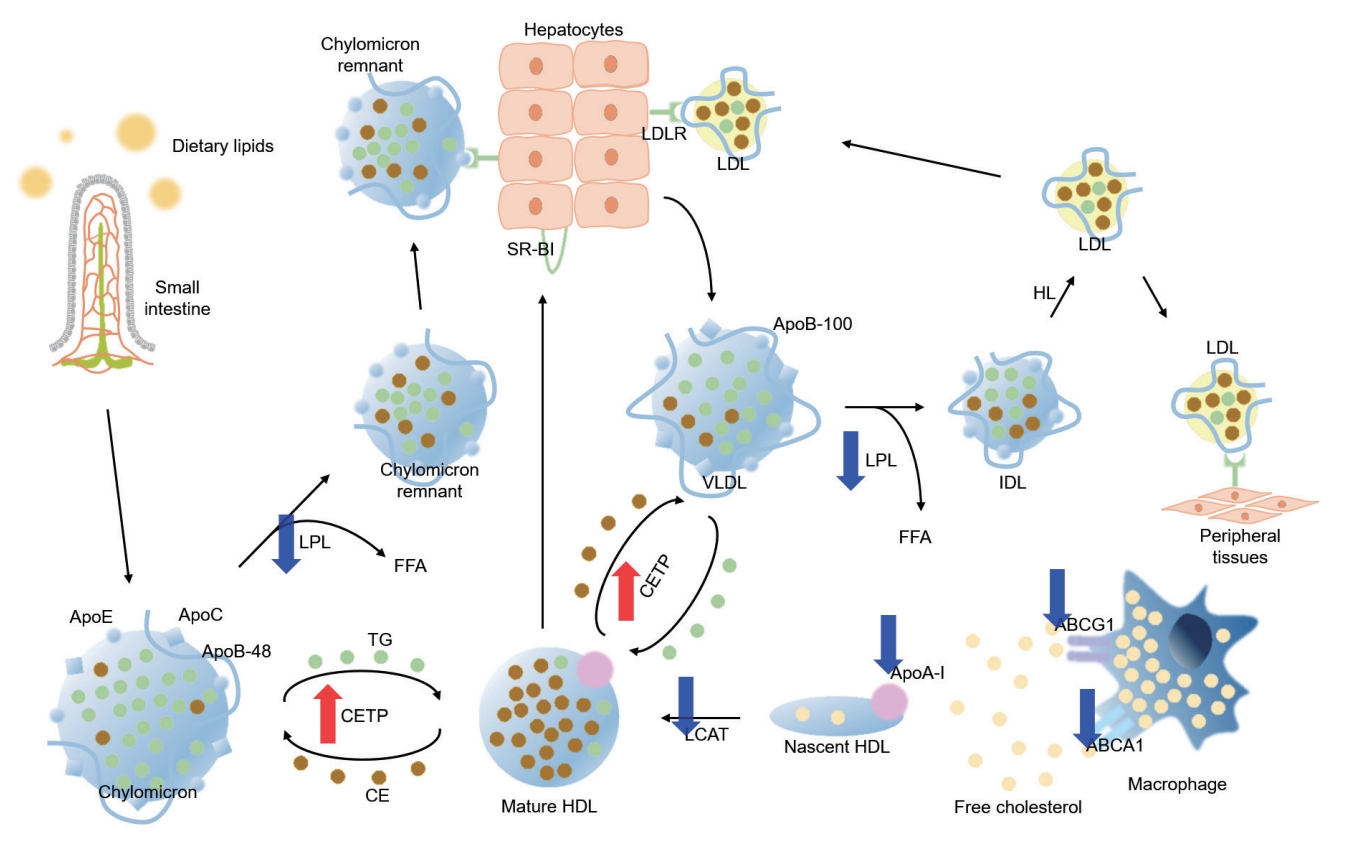
Defective metabolism of major lipoproteins in chronic kidney disease (CKD). Red and blue arrows indicate upregulation/increased activity and downregulation/decreased activity in CKD, respectively. LDLR, low-density lipoprotein receptor; LDL, low-density lipoprotein; SR-BI, scavenger receptor class B type I; LPL, lipoprotein lipase; FFA, free fatty acid; ApoE, apolipoprotein E; ApoC, apolipoprotein C; ApoB, apolipoprotein B; TG, triglyceride; CETP, cholesteryl ester transfer protein; CE, cholesteryl ester; HDL, high-density lipoprotein; HL, hepatic lipase; VLDL, very low-density lipoprotein; IDL, intermediate-density lipoprotein; LCAT, lecithin-cholesterol acyltransferase; ApoA-I, apolipoprotein A-I; ABCG1, ATP-binding cassette subfamily G member 1; ABCA1, ATP-binding cassette subfamily member 1.
Elevated circulating level of lipoprotein(a)
Lipoprotein(a) (Lp(a)) is a unique lipoprotein that has a LDL-like core with a single ApoB molecule linked by disulfide bonds to apolipoprotin(a) (Apo(a)) [46]. Lp(a) particles bind to extracellular matrix and are highly atherogenic [47], such that the rise in Lp(a) levels increased the risk of atherosclerotic cardiovascular disease (ASCVD) in the general population [48]. It is problematic that the use of statins is not effective to lower Lp(a) levels [49], and, rather increase its circulating levels [50], although the precise mechanisms should be further investigated. Elevated circulating level of Lp(a) (usually defined as serum level over 10 to 30 mg/dL) is independently associated with the risk of adverse cardiovascular events also in patients with CKD [1,51]. Moreover, the elevation in Lp(a) levels has been repeatedly reported even with mild reduction in estimated glomerular filtration rate (eGFR) [52,53]. The suggested mechanisms for the elevation in the circulating Lp(a) levels involve the decreased clearance via kidney, based on the finding of a drop in Lp(a) levels in the renal vein compared to the ascending aorta [54]. An animal study in rats demonstrated that injected Lp(a) accumulated in the renal tubules as well as urinary excretion of Apo(a) fragments [55]. On the other hand, some studies suggest that the liver is the major organ for the clearance of Lp(a) [56]. Further studies are warranted to reveal the precise reason for the elevation in the circulating Lp(a) levels.
DYSLIPIDEMIA AND CARDIOVASCULAR OUTCOMES IN RELATION TO CKD
The association between dyslipidemia and the risk of cardiovascular event is best illustrated in the elevation of LDL-C among the general population. In the general population, every 40 mg/dL increase in LDL-C increases the risk of cardiovascular events by 40% [57]. Among the patients on maintenance dialysis, however, this graded relation is largely blunted, or is inversed at below the average level, which is so called ‘reverse epidemiology’ [13,14]. A possible hypothesis for the inverse association between LDL-C levels and all-cause mortality among the patients with end-stage renal disease (ESRD) is that uremic milieu, independently of LDL-C levels, promotes the deaths due to nonatherosclerotic cardiovascular events, such as heart failure and sudden cardiac death, rather that ASCVD [58-60].
Indeed, the patients with ESRD are usually exposed to pathological conditions including anemia, hypervolemia, hypertension and disturbances in bone and mineral metabolism [61-63]. All of these are likely to contribute to the development of uremic cardiomyopathy that is a specific pattern of myocardial fibrosis among the patients with CKD and ESRD [63-65]. A post hoc analysis of the Study of Heart and Renal Protection (SHARP) also supports the hypothesis [66]. The SHARP trial enrolled 9,270 patients with CKD, among which 3,015 patients were on dialysis, and reported a positive association of LDL-C with the risk of major vascular events (hazard ratio [HR], 1.14; 95% confidence interval [CI], 1.06 to 1.22 per 0.6 mmol/L increase in LDL-C), but a negative association with nonatherosclerotic events, including heart failure and arrhythmia (HR, 0.90; 95% CI, 0.83 to 0.97 per 0.6 mmol/L increase in LDL-C) [66].
Albeit high HDL-C levels are protective against cardiovascular events in the general population [6,7], clinical trials failed to demonstrate that pharmacologic interventions, such as niacin, reduce the risk of cardiovascular events [67-70]. While low HDL-C levels are common in patients with CKD, the association between HDL-C and the risk of cardiovascular events is somewhat complicated. In the analyses of the patients with CKD, the association of HDL-C with the risk of cardiovascular events is not any more statistically significant after adjustment for the confounding factors [71,72]. Rather, some studies reported that all-cause mortality is increased with very low or very high levels of HDL-C among the patients with ESRD, probably due to the changes in the composition of lipoproteins related to uremia [73]. It seems like that inflammation modifies the association between HDL-C and cardiovascular risk in patients with nondialysis CKD, as HDL-C levels were inversely associated with the risk of cardiovascular events in the absence of inflammation, whereas HLD-C levels were positively associated with the risk in the presence of inflammation (i.e., high-sensitivity C-reactive protein level ≥1.0 mg/L) [74]. A study reported that non-HDL-C might be a better index for the predication of cardiovascular risk in patients with nondialysis CKD, as a simple, positive correlation was observed between non-HDL-C and the risk of cardiovascular events, even after adjustment for high-sensitivity C-reactive protein level [18].
It has been elucidated that hyperglyceridemia also imposes a residual cardiovascular burden in the general population [5]. Hypertriglyceridemia is also a common phenotype of dyslipidemia in patients with CKD, especially in those with diabetes and those on peritoneal dialysis [75,76]. A recent cohort study of 2.9 million United States veterans including patients with CKD at stages 3a to 5 or ESRD reported a linear association between TG levels and the risk of hospitalization for ASCVD, although the association was attenuated with worsening CKD stages [77]. Interestingly, the study also reported an inverse relationship between TG levels and the risk of non-ASCVD, where elevated TG levels are associated with lower risk, regardless of CKD stages [77].
CARDIOVASCULAR RISK MODIFICATION BY TARGETING DYSLIPIDEMIA IN CKD
Stains
Statins are 3-hydroxy-3-methylglutaryl-coenzyem A reductase inhibitors that primarily target to lower LDL-C levels [78]. Their benefits for the primary [79,80] and secondary [81,82] prevention of cardiovascular events have been robustly established in the general population. Yet, the results from randomized controlled trials (RCTs) including the patients with CKD requires sophisticated precautions to interpret the efficacy of statins to reduce cardiovascular risk.
The Die Deutsche Diabetes Dialyse Studie (4D) trial [83] enrolled 1,255 patients with diabetes who initiated hemodialysis within 2 years, who were randomly assigned to atorvastatin 20 mg daily or placebo. LDL-C levels in those treated with atorvastatin were reduced by 49 mg/dL in average from a median baseline level of 121 mg/dL at 4 weeks, and were persistently maintained for the duration of the study. Despite the significant reduction in LDL-C levels, the incidence of the primary composite outcome (death from cardiac causes, fatal stroke, nonfatal myocardial infarction, and nonfatal stroke) was not significantly different between the two groups during a median of 4 years of follow-up (HR, 0.92; 95% CI, 0.77 to 1.10).
The A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: An Assessment of Survival and Cardiovascular Events (AURORA) trial [84] enrolled 2,776 ESRD patients who had been on hemodialysis for at least 3 months and who were not treated with statins at the baseline. The participants were randomly assigned to rosuvastatin 10 mg daily or placebo. Treatment with rosuvastain decreased LDL-C levels by 43% from a mean baseline level of 100 mg/dL, and decreased C-reactive protein levels as well, although these improvements in the surrogate markers were not accompanied by the reduction in the incidence of primary composite outcome, which is defined as a nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes (HR, 0.96; 95% CI, 0.84 to 1.11).
It should be noticed that the participants in 4D and AURORA trials were not selected by LDL-C levels [83,84]. A post hoc analysis of 4D trial demonstrated atorvastatin significantly reduces the risk of fatal and nonfatal cardiac events and death from any cause among the participants with pretreatment LDL-C level >145 mg/dL [85], although interpretation of secondary analyses from RCTs needs caution, because of reduced statistical power, increased variance, and the play of chance [86,87]. Moreover, cardiovascular events were less specifically defined in 4D and AURORA trials, which also included the deaths from nonatherosclerotic cardiac events [83,84].
The SHARP trial [88] enrolled 9,270 patients with CKD or ESRD to evaluate the efficacy of simvastatin plus ezetimibe for the primary prevention of major atherosclerotic event, which was defined as nonfatal myocardial infarction or coronary death, nonhemorrhagic stroke, or arterial revascularization excluding dialysis access procedures. Contrary to the previous two RCTs, 6,247 patients were nondialysis-dependent, and the remaining 3,023 patients were on maintenance dialysis, where overall 75% of the study participants had CKD stages at 4 to 5 or ESRD. The patients were initially randomly assigned to simvastatin plus ezetimibe, simvastatin alone, and placebo groups. Those who were initially assigned to simvastatin alone group received simvastatin plus ezetimibe 1 year after the initiation of the study. Treatment with simvastatin plus ezetimibe yielded an average reduction of LDL-C level by 33 mg/dL. During a median follow-up of 4.9 years, simvastatin plus ezetimibe significantly reduced the risk of major atherosclerotic events (HR, 0.83; 95% CI, 0.74 to 0.94).
Meta analyses report a clear risk reduction of cardiovascular event by statin therapy in the general population [89], while the effect is substantially modified in patients with CKD [60,90], indicating that the risk reduction of cardiovascular event per reduction in LDL-C levels is attenuated as CKD progresses [90], with no detectable benefits in patients with ESRD conferred by statin therapy [60]. This is also consistently well-demonstrated in the individual RCTs, such as 4D, AURORA, and SHARP trials [83,84,88]. The possible explanations have been suggested. First, a considerable portion of cardiovascular events in patients with ESRD may be due to nonatherosclerotic pathology, such as sudden cardiac death and heart failure [58,59], which could not be essentially aimed by statin therapy. Second, the number of the participants with ESRD might have been not sufficient to demonstrate the statistical significance, compared to the other studies with positive results, such as the Scandinavian Simvastatin Survival Study (n=4,444) [79] and the West of Scotland Coronary Prevention Study (n=6,595) [91]. Third, the effect of statins on the elevation of circulating Lp(a) levels may be maximized in patients with ESRD. It is known that statins tend to increase circulating Lp(a) levels to contribute to the residual cardiovascular burden in the general population [92]. As Lp(a) is cleared via kidney, its level is usually highest in patients with ESRD [48]. Therefore, we cannot exclude the possibility that the effect of statins on the elevation of circulating Lp(a) levels may be maximized in patients on dialysis to null the cardioprotective effect by LDL-C lowering, although the hypothesis remains to be further validated.
Based on the observations from RCTs, the 2013 KDIGO clinical practice guideline for lipid management in CKD [11] and the others [8-10] commonly do not recommend the initiation of statin therapy in patient on dialysis who are not being treated with statins, while the continuation of statin therapy is acceptable for those who are already on statin therapy. In this context, a retrospective analysis of United States veterans initiating dialysis (n=14,298) reported that the continuation of statin therapy was associated with reduced all-cause mortality (HR, 0.72; 95% CI, 0.66 to 0.79) and cardiovascular mortality (HR, 0.82; 95% CI, 0.69 to 0.96) [93]. The administration of statins among the indicated patients with nondialysis CKD is a widely accepted concept throughout current guidelines, while varying targets for LDL-C levels are still on the controversy (Table 1) [8-11,94].

Recommendations from selected guidelines for LDL-C lowering therapy in patients with CKD
Statin therapy in kidney transplant recipients to lower LDLC levels is also usually advocated by the current guidelines [9-11], although the class of recommendation is not strong. The Assessment of Lescol in Renal Transplantation (ALERT) study [95] enrolled 2,102 kidney transplant recipients receiving cyclosporine-based immunosuppression with stable graft function, and randomly assigned the participants to fluvastatin 40 mg daily or placebo. During a median follow-up of 5.1 years, LDL-C levels were decreased by 40 mg/dL in the intervention group, with a significant reduction in nonfatal myocardial infarction and cardiac deaths, although the incidence of the primary composite outcome of major cardiac events (defined as cardiac death, nonfatal myocardial infarction, or coronary intervention) was not significantly different between the two groups. The benefits of fluvastatin therapy in the initial study in terms of nonfatal myocardial infarction and cardiac death were sustained, even when all of the participants were offered open-label, long-term high dose of fluvastatin (80 mg) during a complex extension study period for a total of 6.7 years [96]. The results from the RCTs of statin therapy in patients with CKD are summarized in Table 2.

Randomized controlled trials of statin therapy in patients with chronic kidney disease
Fibrates
Fibrates stimulate peroxisome proliferator-activated receptor alpha (PPARα) to effectively reduce TG levels and elevate HDL levels. RCTs have demonstrated the efficacy of fibrates in the primary and secondary prevention of cardiovascular events in the general population. For instance, in Helsinki Heart Study, treatment of gemfibrozil in asymptomatic men with non-HDL-C ≥200 mg/dL significantly increased HDL-C and reduced total and LDL-C, and TG levels. Importantly, during the follow-up period of 5 years, the incidence of coronary heart disease was reduced by 34% in the gemfibrozil group compared to the placebo group. Considering the common feature of dyslipidemia in patients with CKD, fibrates should be an attractive therapeutic option. Currently, there are no RCTs available that directed included the patients with CKD to examine the cardioprotective effects of fibrate therapy. A meta-analysis, however, identified a total of 16,869 participants with CKD from RCTs that assess the effects of fibrate therapy compared with placebo in people with CKD, and reported that fibrates improve lipid profiles and prevent cardiovascular events in the subjects with eGFR <60 mL/min/1.73 m2 as well as in the subjects with eGFR <30 mL/min/1.73 m2 [97]. Whereas the use of fibrates is associated with increased serum creatinine levels especially among those with eGFR <30 mL/min/1.73 m2, a recent meta-analysis reported that initial increase in creatinine remains relatively constant afterwards, suggesting the use of fibrates does not significantly accelerate the progression of CKD [98]. Although 2013 KDIGO clinical practice guideline for lipid management in CKD recommends therapeutic lifestyle modification in the adults with CKD and hypertriglyceridemia, primarily due to lack of no published RCTs in CKD populations and too few participants with CKD included in previous trials to provide reliable information, it should be reminded that the recommendation do not entirely exclude the beneficial effect of fibrates in selected individuals with CKD.
Omega-3 fatty acid
Omega-3 fatty acid (eicosapenatnoic acid [EPA] and docosahexenoic acid) at pharmacologic dose (2 to 4 g/day) reduce TG levels by up to 45%, with little effect on LDL-C or HDL-C levels, in the general population as well as in patients with CKD, although the action mechanism is poorly understood. The effect of omega-3 fatty acid added on statin therapy for the reduction of residual cardiovascular burden is controversial. The Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial (REDUCE-IT) was designed to address the efficacy of icosapent ethyl (IPE), a highly purified EPA ethyl ester on the risk of cardiovascular events among patients with elevated TG levels despite the use of statins. The primary endpoint was the composite of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, coronary revascularization, or unstable angina requiring hospitalization. The participants were followed for a median of 4.9 years. The result demonstrated that the risk of cardiovascular event is significantly decreased with medication of IPE 2 g twice daily (HR, 0.71; 95% CI, 0.59 to 0.85) [99]. In a post hoc analysis of REDUCE-IT study, where the participants (n=8,179) were categorized by eGFR into eGFR ≥90 mL/min/1.73 m2, eGFR 60 to <90 mL/min/1.73 m2 and eGFR <60 mL/min/1.73 m2 (the median baseline eGFR 75 mL/min/1.73 m2 [range, 17 to 123]), the benefits of IPE on primary and key secondary composite endpoints were consistently observed across the eGFR categories, albeit the effect was less clear among the patients with eGFR <45 mL/min/1.73 m2 [100]. The Long-Term Outcomes Study to Assess Statin Residual Risk with Epanova in High Cardiovascular Risk Patients with Hypertriglyceridemia (STRENGTH) trial reported a contrary result [101]. The STRENGTH trial intended to test the efficacy of a carboxylic acid formulation of EPA (omega-3 CA) in patients with CKD in statin-treated participants (n=13,078) with high cardiovascular risk, hypertriglyceridemia, and low levels of HDL-C. The participants were randomly assigned to receive 4 g/day of omega-3 CA or corn oil, in addition to statins. The primary endpoint was the composite of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, coronary revascularization, or unstable angina requiring hospitalization. During the median follow-up duration of 42 months, the addition of omega-3 CA, compared with corn oil, resulted in no significant difference in a composite outcome of cardiovascular events (HR, 0.99; 95% CI, 0.90 to 1.09). Moreover, a high rate of new-onset atrial fibrillation was observed with the use of omega-3 CA than corn oil (2.2% vs. 1.3%, P<0.001). Currently there is no plausible explanation for the obviously discrepant results from the two RCTs. Although the Food and Drug Administration approved the use of IPE for the prevention of cardiovascular events based on the results from REDUCE-IT study, and the 2019 ESC/EAS guidelines for the management of dyslipidemias recommend to consider the use of IPE in high-risk patients with hypertriglyceridemia despite statin treatment [102], molecular targets of IPE have not been identified yet. Moreover, the cardiovascular risk reduction observed in REDUCE-IT study was not associated with the magnitude of the reduction in TG levels. The precise action mechanism of IPE should be further elucidated.
Proprotein convertase subtilisin/kexin type 9 inhibitors
Binding of proprotein convertase subtilisin/kexin type 9 (PCSK9), a secreted serine protease, to ectodomain of low-density lipoprotein receptor (LDLR) on the surface of hepatocytes promotes lysosomal degradation of LDLR, which in turn reduces the clearance of LDL particles from the circulation, resulting in increased LDL-C levels [103,104]. Monoclonal antibodies targeeting PCSK9, such as alirocumab [105-115] and evolocumab [116-120], are currently available as PCSK9 inhibitors, which effectively lower LDL-C levels even among those who were already on statin therapy, and reduced the risk of adverse cardiovascular events. Contrary to statins, the data on PCSK9 inhibitors in patients with CKD is very limited, because RCTs on PCSK9 inhibitors mostly excluded the patients with eGFR <20 to 30 mL/min/1.73 m2 [105-120]. An analysis of the pooled data from eight phase III the Effect of Alirocumab on the Occurrence of Cardiovascular Events in Patients Who Have Recently Experienced an Acute Coronary Syndrome (ODYSSEY) trials included a total of 4,629 patients with hypercholesterolemia, among whom 467 patients had CKD stage 3, which demonstrated that alirocumab consistently lowers LDL-C levels regardless of impaired renal function, although the patients with eGFR <30 mL/min/1.73 m2 were excluded from the analysis [121]. In a post hoc analysis of the Further cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) trial, 8,077 patients with preserved kidney function (eGFR ≥90 mL/min/1.73 m2), 15,034 with CKD stage 2 (eGFR 60 to <90 mL/min/1.73 m2), and 4,443 with eGFR <60 mL/min/1.73 m2 were included to analyze the efficacy and safety of evolocumab in patients with CKD. Similar to the result from ODYSSEY trials, the efficacy of lowering LDL-C levels safety of evolocumab vs. placebo were consistent across subgroups defined by different eGFR range ([HR, 0.82; 95% CI, 0.71 to 0.94] in patients with preserved renal function; [HR, 0.85; 95% CI, 0.77 to 0.94] in patients with stage 2 CKD; [HR, 0.89; 95% CI, 0.76 to 1.05] in patients with stage 3 CKD; P for interaction=0.77) [122]. The patients with eGFR <30 mL/min/1.73 m2 were excluded from FOURIER trial. Whereas the potent lipid lowering effect of PCSK9 inhibitors should be attractive, it remains to be further evaluated whether PCSK9 inhibitors can be safely administered and ultimately reduce the risk of adverse cardiovascular events in patients with advanced CKD (eGFR <20 to 30 mL/min/1.73 m2).
DYSLIPIDEMIA AND CKD PROGRESSION
Although the management of dyslipidemia primarily targets the reduction of the risk for cardiovascular events, the parameters related to kidney function, such as eGFR decline, proteinuria, and initiation of dialysis, are also the clinical outcomes of particular importance in patients with CKD. Given that the kidney is a highly vascularized organ, and that the overall effect of dyslipidemia should be also deleterious in the vascular bed of the kidney, it is readily expectable that adverse renal outcomes might be associated with dyslipidemia in patients with CKD. In this regard, mounting evidence suggests the association between various abnormalities in the lipid profile and CKD progression, which are mostly derived from cohort studies.
For instance, elevated LDL-C levels seem to be associated with the risk of CKD progression [15]. In the analysis of 1,886 patients with CKD stages 1 to pre-dialysis 5, the risk of composite kidney event (defined as a composite of 50% decline in eGFR during follow-up or onset of end-stage kidney disease including initiation of dialysis or kidney transplantation) was highest in those with LDL-C ≥130 mg/dL (HR, 2.05; 95% CI, 1.19 to 3.56) compared to those with LDL-C <70 mg/dL during the median follow-up of 5.2 years [15]. Hypertriglyceridemia is also associated with the progression of CKD [16]. In a prospective cohort study of the patients with nondialysis CKD at stage 1 to 5, a total of 2,158 participants were divided into the quartile by serum TG levels, where, compared to the 1st quartile (TG <92 mg/dL), the risk of CKD progression was significantly higher (HR, 1.43; 95% CI, 1.05 to 1.96) in the 4th quartile (TG ≥194 mg/dL) during the median follow-up of 6.9 years [16]. The association of low HDL-C levels with the risk of CKD progression is somewhat complicated. In a study that evaluated the association between serum HDL-C levels and the risk of CKD progression in 2,168 patients with nondialysis CKD, the risk of CKD progression was lowest in the reference group with with HDL-C levels of 50 to 59 mg/dL, whereas both low (<30 mg/dL: HR, 2.21; 95% CI, 1.30 to 3.77) or high (≥60 mg/dL: HR, 2.05; 95% CI, 1.30 to 3.77) HDL-C levels were associated with significantly increased risk of CKD progression during a median follow-up of 3.1 years, suggesting a U-shaped association between serum HDL-C levels and adverse renal outcomes [17]. A study from the same cohort that investigated the association between non-HDL-C levels and the risk of CKD progression also reported a U-shaped association [19].
Accordingly, reports focused at improving the renal outcomes by correction of dyslipidemia followed, although the most were the results from the secondary analysis of RCTs or cohort studies. The effect of lowering LDL-C by statins on the delaying of CKD progression is disappointing. In the post hoc analysis of SHARP trial that included a total of 6,245 patients with nondialysis CKD, the incidence of ESRD was not significantly reduced in the simvastatin plus ezetimibe group compared to the placebo group (rate ratio, 0.97; 95% CI, 0.89 to 1.05) during a median follow-up of 4.8 years [123]. Even in a meta-analysis published in 2016, which included 143,888 participants from seven RCTs concluded that, despite the modest reduction in the proteinuria and the rate of eGFR decline, the risk kidney failure event, defined as a composite of more than 25% or 50% decrease in eGFR, doubling of serum creatinine level, or onset of ESRD, was not ultimately reduced by statin therapy (odds ratio, 0.98; 95% CI, 0.87 to 1.10) [124]. The protective effect of statins against CKD progression was denied even in a cohort study, which reported that intensity of statin therapy is not significantly relevant to the risk of CKD progression, suggesting that long-term kidney outcomes may not be modified by statin therapy [125].
The effect of pharmacologic interventions other than statins on the CKD progression is largely negative or unknown due to lack of data. In a clinical trial to test the effect of omega-3 fatty acids on the development or progression of CKD in patients with type 2 diabetes mellitus, the rate of eGFR decline for 5 years was not significantly reduced in the omga-3 fatty acid group compared to the placebo group, regardless of vitamin D3 supplement [126]. The role of fibrates in the delaying of CKD progression, however, remains to be further tested. In a meta-analysis of the clinical trial that evaluated the clinical benefits and safety of fibrate therapy, the risk of albuminuria progression was significantly reduced by fibrate therapy (risk ratio, 0.86; 95% CI, 0.76 to 0.98), although the risk of eGFR reduction was also significantly increased with fibrate therapy (mean difference, –2.67 mL/min/1.73 m2; 95% CI, –4.81 to –0.54) [97]. Yet, as previously mentioned, the use of fibrates does not seem to significantly accelerate the progression of CKD, as the initial increase in creatinine remains relatively constant afterwards [98]. Moreover, a recent nation-wide cohort study conducted in Taiwan, which only included that the patients with advanced CKD, reported that monotherapy with fenofibrate was associated the lowest incidence of permanent dialysis (fenofibrate vs. nonuser: subdistribution hazard ratio [SHR], 0.78; 95% CI, 0.77 to 0.80; statins vs. fenofibrate: SHR, 1.27; 95% CI, 1.26 to 1.29; combination of statins and fenofibrate vs. fenofibrate: SHR, 1.15; 95% CI, 1.13 to 1.17) [127]. It is impressive that the index date of ‘advanced CKD’ in this study was operationally defined as the first prescription of an erythropoiesis-stimulating agent, which can be used in the case of eGFR <15 mL/min/1.73 m2 and a hematocrit level <28% in Taiwan, suggesting the potential efficacy of fenofibrate to delay the progression of CKD even in advanced stages. Currently, a clinical trial to address the effect of pemafibrate, a novel selective PPARα modulator, on the kidney protection in patients with CKD in on-going [128]. The key eligible criteria of the Pemafibrate, open-label, Randomized cOntrolled study to evaluate the renal protective eFfect In hypertriglyceridemia patients with Chronic Kidney Disease (PROFIT-CKD) study include spot urine protein-to-creatinine ration ≥0.15 g/gCr within 3 months before enrollment and fasting TG ≥150 mg/dL and <1,000 mg/dL at enrollment. It is expected that the study may present a conclusive result to determine the overall effect of fibrates on the kidney function in patients with CKD, as the estimation of eGFR at the baseline and 12-month follow-up is scheduled as a secondary endpoint, although those with serum creatinine ≥2.5 mg/dL or creatinine clearance <40 mL/min are excluded.
CONCLUSIONS
The management of dyslipidemia in CKD is obviously challenging. Besides the lack of high-quality evidence derived from CKD population, the impact of dyslipidemia on cardiovascular outcomes seems to be substantially modified as kidney function deteriorates. Further, it has been questionable whether the unique pattern of alterations in lipid profile of CKD population should be the target of therapy. Based on all of these contexts, it seems natural that the 2013 KDIGO guideline presented a relatively conservative approach with respect to the indication of lipid lowering therapy and therapeutic monitoring among the patients with CKD. However, the currently available data suggests that elevated LDL-C levels still increase the risk of atherosclerotic adverse events even in patients with ESRD, and that hypertriglyceridemia also impose residual cardiovascular burden in this population. A sophisticated analysis of updated evidence suggests that the use of statins, omega-3 fatty acid or fibrates might be beneficial on the basis of the individualized assessment of the risk for cardiovascular disease even among those who are not routinely indicated in the guidelines. The use of PCSK9 inhibitors should also be expanded in patients with CKD, provided that evidence for the patients with CKD will be accumulating. Moreover, there is compelling evidence that dyslipidemia is associated with CKD progression. Although the effect of statins to delay CKD progression by lowering LDL-Cs have been consistently failed, the role of pharmacologic interventions other than statins on the CKD progression is largely undetermined yet. In this regard, targeting hypertriglyceridemia by fibrates may be promising to prevent CKD progression, while conclusive results are being awaited from RCTs.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
This work wassupported by the National Research Foundation of Korea (NRF) funded by the Korea government (MSIT) (RS2023-00217317, NRF -2020R1F1A1074001) and a grant (BCRI 22042 and BCRI22079) of Chonnam National University Hospital Biomedical Research Institute.
Acknowledgements
None