Endoplasmic Reticulum Stress and Dysregulated Autophagy in Human Pancreatic Beta Cells
Article information

Abstract
Pancreatic beta cell homeostasis is crucial for the synthesis and secretion of insulin; disruption of homeostasis causes diabetes, and is a treatment target. Adaptation to endoplasmic reticulum (ER) stress through the unfolded protein response (UPR) and adequate regulation of autophagy, which are closely linked, play essential roles in this homeostasis. In diabetes, the UPR and autophagy are dysregulated, which leads to beta cell failure and death. Various studies have explored methods to preserve pancreatic beta cell function and mass by relieving ER stress and regulating autophagic activity. To promote clinical translation of these research results to potential therapeutics for diabetes, we summarize the current knowledge on ER stress and autophagy in human insulin-secreting cells.

INTRODUCTION
Pancreatic beta cell function and mass are important in the pathogenesis and treatment of diabetes because insulin hormone released from beta cells is critical for glucose homeostasis. Pancreatic islets and beta cell lines from rodents have been usual materials for beta cell research because human beta cells were difficult to obtain. However, human islets differ from rodent islets in several ways, including their architecture, metabolism, turnover rate, and plasticity [1–6]. Although animal models have contributed to our understanding of human physiology and disease, they cannot accurately predict efficiency and toxicity in drug discovery. The high failure rate of candidate compounds in clinical trials can be explained, at least in part, by the limitation of extrapolating animal findings to human [7]. Considering the substantial inter-species differences in pancreatic islets between rodents and humans, proof-of-concept studies using cells of human origin are essential for clinical translation.
Several centers and consortia in Europe and the USA have isolated human islets from deceased organ donors for research. In addition, human beta cell lines were generated using genetic oncogenesis and electrofusion with a pancreatic adenocarcinoma cell line [8,9]. Engineering stem cells into beta cell-like cells has also been reported [10]. These should increase the use of human-originated insulin-producing cells in research, although the resources still have some limitations.
In rodent beta cells, endoplasmic reticulum (ER) stress and dysregulated autophagy were shown to be inter-related features in diabetes and potential therapeutic targets [11–14]. Thus, it is time to summarize the findings in human islets, beta cell lines, and beta cell-like cells, which are covered in this mini-review, where a brief description of current knowledge in non-human cells precedes.
ER STRESS IN BETA CELLS AND DIABETES
ER stress and the unfolded protein response
The ER is a crucial intracellular organelle involved in protein synthesis, transport, folding, and degradation. ER senses accumulating misfolded or unfolded proteins, which lead to ER stress and activates a signaling pathway termed the unfolded protein response (UPR). The UPR is mediated by transmembrane proteins in the ER including protein kinase R-like ER kinase (PERK or eukaryotic translation initiation factor 2-alpha kinase 3 [EIF2AK3]), inositol-requiring protein-1 (IRE1), and activating transcription factor-6 (ATF6) (Fig. 1). Under ER stress, activated PERK phosphorylates eukaryotic translation initiation factor 2 alpha (eIF2A), which downregulates general protein translation to alleviate the ER burden, resulting in adaptive UPR. Persistent ER stress and an excessive UPR upregulates the expression of activating transcription factor-4, leading to ER stress-induced apoptosis by activating DNA damage-inducible transcript 3 (DDIT3/C/EBP homologous protein [CHOP]/growth arrest-and DNA damage-inducible gene 153 [GADD153]), which is called maladaptive UPR. Another ER stress sensor, IRE1, is auto-phosphorylated under ER stress, which promotes the expression of UPR target genes that encode ER chaperones and proteins related to folding, trafficking, and transport. Under ER stress, ATF6 is transported to the Golgi apparatus and then to the nucleus, which induces the expression of UPR target genes. The detailed mechanisms of the UPR have recently been reviewed [15].

Endoplasmic reticulum (ER) stress in pancreatic beta cells. In vitro, ex vivo, and in vivo human findings are depicted in red, along with the references. TIDM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; MODY, maturity onset diabetes of the young; IL, interleukin; WFS1, wolframin ER transmembrane glycoprotein; BIP, binding immunoglobulin protein; IRE1, inositol-requiring protein-1; PERK, protein kinase R-like ER kinase; ATF6, activating transcription factor-6; EIF2B1, eukaryotic translation initiation factor 2B subunit alpha; eIF2A, eukaryotic translation initiation factor 2-alpha; XBP1, X-box binding protein 1; ATF4, activating transcription factor-4; UPR, unfolded protein response; CHOP, C/EBP homologous protein; ATF6f, cytoplasmic fragment of activating transcription factor-6; TUDCA, tauroursodeoxycholic acid.
ER stress in pancreatic beta cells and therapeutic approaches
The capacity to regulate misfolded or unfolded proteins in the ER varies among cell types. Pancreatic beta cells have a larger and more highly developed ER than other cells, because they need to synthesize and secrete large quantities of insulin molecules in a short time [16]. Under diabetic conditions, high glucose levels excessively stimulate beta cells and increase proinsulin synthesis, which exceeds the capacity of the ER to eliminate misfolded proteins, leading to ER stress and beta cell apoptosis [11].
Proinsulin misfolding was observed in the islets of leptin receptor-deficient diabetic mice, as the formation of aberrant disulfide-linked proinsulin complexes. These Cys(B19)-Cys(B19) covalent proinsulin homodimers resist reductive dissociation, highlighting the structural basis of aberrant proinsulin complex formation. Because the further increase in these complexes tracks with the onset of islet insulin deficiency in genetically obese mice with wild-type islets, the formation and accumulation of aberrant disulfide-linked proinsulin complexes was postulated as an early event in type 2 diabetes mellitus (T2DM) [17].
Chronic hyperglycemia also enhances the generation of reactive oxygen species (ROS) via oxidative phosphorylation overload in mitochondria. Excessive ROS induces oxidative and ER stress [18]. Additionally, prolonged elevation of free fatty acid (FFA) levels triggers ER stress in beta cells. Palmitate-induced lipotoxicity activates the UPR signaling pathway via ER stress sensors, which leads to attenuation of new protein synthesis and induction of ER chaperones and beta cell apoptosis [19,20].
Disturbed ER homeostasis and unmitigated ER stress are believed to lead to beta cell failure, which can be prevented by the adaptive UPR. Meanwhile, a maladaptive UPR also induced apoptosis (Fig. 1); therefore, specific inhibitors of the UPR were developed to prevent an excessive UPR and cell death. Blocking the PERK branch with an inhibitor (GSK2606414 or GSK2656157) efficiently reduced a maladaptive UPR and had therapeutic effects against malignancy and neurodegeneration in mouse models [21,22]. Unfortunately, in beta cells, the PERK inhibitors abrogated glucose-stimulated insulin secretion (GSIS) and calcium dynamics [23] and deregulated protein synthesis, leading to rapid accumulation of misfolded proinsulin and ER stress [24]. Resultantly, in vivo PERK inhibitors induced hyperglycemia in mice via islet damage [21,22]. In contrast, modulating the IRE1 branch in beta cells using small molecules ATP-competitive IRE1α kinase inhibiting RNase attenuator 6 (KIRA6) and KIRA8 increased beta cell survival and function to improve hyperglycemia in insulin deficient diabetic mice such as Akita mice and non-obese diabetic (NOD) mice [25,26]. These KIRAs ameliorated ER stress-induced hyperactivation and endoribonuclease activity of IRE1α and degradation of ER-localized mRNAs. Evidence for the pharmacologic targeting of ER stress and plausible mechanisms have been reviewed elsewhere [11,12,27,28].
Observations in human cells
Accumulating evidence from studies using human-origin cells shows that balancing ER homeostasis in beta cells is essential for maintaining their function. Diabetes-induced ER stress contributes to beta cell failure. Attenuating ER stress has therapeutic effects in diabetes (Fig. 1).
Monogenic diabetes suggests ER stress as a pathogenic factor
Wolcott-Rallison syndrome, a rare autosomal recessive disorder by loss-of-function mutations in PERK causes multi-organ defects and early onset diabetes, mediated by ER stress: apoptosis of beta cells caused by ER stress due to an insufficient UPR leads to permanent neonatal diabetes mellitus [29]. Missense mutations in EIF2B1 also cause permanent neonatal diabetes mellitus, presumably due to a failure of eIF2B to sense eIF2 phosphorylation, leading to an unregulated UPR [30]. Wolfram syndrome is an autosomal recessive disorder caused by mutations in encoding wolframin ER transmembrane glycoprotein (WFS1), resulting in beta cell dysfunction via an ER stress-dependent mechanism [31].
Maturity onset diabetes of the young (MODY) type 10, also known as mutant insulin gene-induced diabetes of youth (MIDY), results from heterozygous mutations in INS. Not all mutations in INS cause this autosomal dominant disorder, but most missense mutations that affect proinsulin folding induce MIDY, along with accumulation of the misfolded proinsulin in the ER, suggesting that perturbations of proinsulin folding exert ER stress and induce beta cell demise [32]. In a patient-derived induced pluripotent stem cell (iPS) model of neonatal diabetes, INS mutations that interrupt proinsulin translation rather reduced ER stress [33]. Collectively, both balanced proinsulin synthesis and structural modifications are needed to escape ER stress in beta cells.
Type 1 diabetes mellitus-associated ER stress
An autoimmune response has been suggested to drive ER stress in type 1 diabetes mellitus (T1DM) [34]. An increase in the expression of ER stress markers, such as CHOP, was detected in the islet cells of patients with T1DM [35]. The proinsulin-to-C-peptide ratio, a clinical measure of ER dysfunction in beta cells, is also pathologically elevated [36]. A T1DM-associated proinflammatory cytokine mix of interleukin (IL)-1β, tumor necrosis factor-α, and interferon-γ induced ER stress and apoptosis via activation of mitogen-activated protein kinase 8 in human beta cells. Tauroursodeoxycholic acid (TUDCA), a chemical chaperone, can alleviate the cytokine-induced apoptosis [37].
T2DM-associated ER stress
Insulin resistance is involved in the progression to T2DM. Beta cells adapt to insulin resistance by increasing insulin production. Insulin biosynthesis begins with the folding of proinsulin, including the formation of three disulfide bonds. Normal pancreatic islets contain a subset of proinsulin molecules bearing at least one free cysteine thiol; under perturbed ER homeostasis, induced with the PERK inhibitor GSK2606414, this non-native proinsulin enters intermolecular disulfide-linked complexes, leading to aggregates of misfolded proinsulin in the ER [17]. Data from both rodent models and patients with MODY type 10 suggest that the aggregates of misfolded proinsulin increase ER stress and trigger insulin deficiency, and these proinsulin aggregates contribute to the development/aggravation of T2DM. Elevation of circulating proinsulin or the proinsulin-to-C-peptide ratio is predictive of progression to T2DM [38].
Glucolipotoxicity-induced ER stress in vitro
Chronic elevation of blood glucose and saturated fatty acid levels is prominent in T2DM. However, whether these elevations actually cause beta cell toxicity, so-called glucolipotoxicity, in humans is a matter of debate [39,40]. Over the past decade, studies using omics techniques with human islets have provided evidence that prolonged exposure to high levels of glucose and saturated fatty acids induces genetic changes relevant to islet dysfunction in T2DM as well as epigenetic changes related to insulin secretion in a time-dependent manner, with cell-protective signals increasing during the earlier period and then decreasing later [41–45]. This is compatible with the finding that acute exposure to FFAs exerts a stimulatory effect on insulin secretion, which eventually deteriorates [44,46].
RNA sequencing analyses of human islets exposed to 0.5 mM palmitic acid for 48 hours identified novel mediators of adaptive ER stress signaling, including palmitate-modified genes regulating ubiquitin and proteasome function, autophagy, and apoptosis. Inhibition of autophagic flux and lysosomal function contributes to lipotoxicity [47]. One striking difference between human and rodent diabetes models is that deposition of amyloid in the pancreatic islets occurs only in humans [48]. Human islet amyloid polypeptide (IAPP) is co-secreted with insulin and is prone to forming extracellular oligomer plaques in islets. FFAs induce IAPP expression in human beta cells, and IAPP aggregation contributes to ER stress [47,49]. One important role of autophagy is removal of aggregated IAPP from beta cells, which will be further discussed in Section 2.
ER stress in beta cells as a therapeutic target
Modulation of the UPR to expand beta cell mass might have therapeutic potential in people at risk for diabetes, because the UPR was necessary and sufficient to increase the proliferation of human beta cells via the ER stress sensor ATF6 [50]. Several cytokines, such as IL-22 and IL-10, alleviated ER stress in human islets initiated by either cytokines or palmitic acid [51].
Proof-of-concept trials using repurposed therapies have been performed in patients with T1DM. A preclinical investigation in mice suggested that the tyrosine kinase inhibitor imatinib might act, at least in part, by counteracting the high levels of ER stress in beta cells and reducing apoptosis, because imatinib inhibits both the colocalization of c-Abl with IRE1α at the ER membrane and IRE1α hyperactivation [26]. A multicenter, randomized, double-blind, placebo-controlled, phase II trial of imatinib demonstrated preserved beta cell function in patients with recent-onset T1DM. During imatinib therapy, the proinsulin to C-peptide ratio was low, which has been linked to reduced ER stress [52]. Another phase II trial of TUDCA in patients with T1DM is ongoing (NCT02218619). TUDCA is a taurine-conjugated bile acid and was identified as a chemical chaperone that alleviates ER stress and reduces diabetes in NOD mice.
AUTOPHAGY IN BETA CELLS AND DIABETES
Physiologic role of autophagy in cellular homeostasis
Autophagy is classified into three major types: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy is often simply referred to as autophagy, and in this review, autophagy refers to macroautophagy. When autophagy is initiated, an isolation membrane forms in the cytosol, which is gradually elongated via two pathways of ubiquitin-like conjugation to form an enclosed mature autophagosome, a cellular cargo containing debris and damaged organelles. Autophagosomes eventually fuse with lysosomes to form autophagolysosomes, which are then degraded by lysosomal enzymes and recycled (Fig. 2) [53]. This process is crucial for maintaining quality of cellular components, functions, and survival. However, like the effect of an excessive UPR on ER stress, excessive activation of autophagy can induce autophagic cell death, also called type II cell death [54].
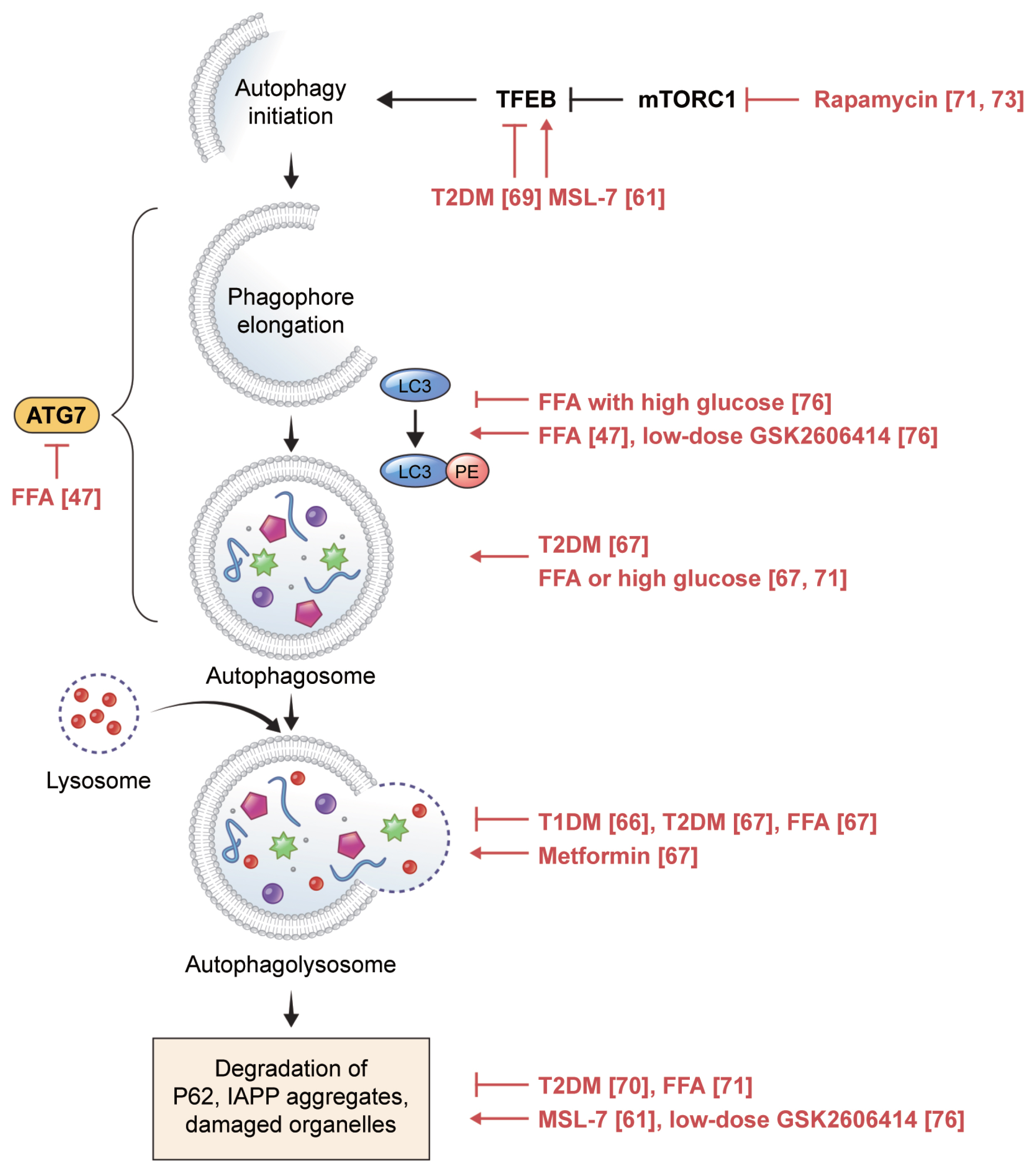
Autophagic process in pancreatic beta cells. In vitro and ex vivo human findings are depicted in red, along with the references. TFEB, transcription factor EB; mTORC1, mTOR complex I; T2DM, type 2 diabetes mellitus; MSL-7, autophagy enhancer; ATG7, autophagy-related 7; LC3, microtubule-associated protein 1 light chain 3; FFA, free fatty acid; PE, phosphatidylethanolamine; T1DM, type 1 diabetes mellitus; IAPP, islet amyloid polypeptide.
Dysregulated autophagy in pancreatic beta cells and therapeutic approaches
Impaired autophagy in pancreatic beta cells has been implicated in the development of diabetes. Genetic inhibition of autophagy affected organelles in beta cells, such as the mitochondria and ER, and reduced beta cell mass and insulin secretion, resulting in hyperglycemia in mice with and without obesity [55,56].
Amyloid accumulation in pancreatic islets is a feature of T2DM that is not observed in rodents because mouse IAPP (mIAPP) does not generate aggregates. However, in transgenic mice with beta cell-specific expression of human IAPP (hIAPP), a deficiency in autophagy resulted in the development of overt diabetes, which was not observed in mice expressing hIAPP alone or with an autophagy deficiency alone. Lack of autophagy in hIAPP-expressing mice results in amyloid accumulation in pancreatic islets, leading to increased beta cell death [57–59]. Therefore, autophagy plays a role in defending beta cells against amyloid-induced toxicity. MSL-7, an autophagy enhancer identified by high-throughput screening of a chemical library against metabolic syndrome, was reported to have virtually no effect on the beta cells of high-fat diet-fed mice [60]. However, MSL-7 significantly reduced the accumulation of hIAPP in the islets of hIAPP-expressing mice fed a high-fat diet, thereby improving beta cell function and hyperglycemia [61].
ER-phagy, or reticulophagy, is a type of selective autophagy that reduces ER load via lysosomal degradation of excess or damaged ER. ER stress triggers ER-phagy, and impaired ER-phagy increases ER stress; therefore, crosstalk between these organelles contributes to beta cell health [62,63]. In Akita mice with a mutation in the Ins2 gene, misfolded proinsulin accumulates in ER aggregates with wild-type proinsulin, resulting in enhanced ER stress and impaired insulin secretion. According to Cunningham et al. [64], the ER membrane protein reticulon 3 (RTN3) mediated ER-phagy and eliminated the aggregates of Akita proinsulin transfected in INS-1 cells. However, whether Akita proinsulin-associated ER stress induces RTN3 has not yet been determined. The detailed mechanisms of ER-phagy induced by ER stress in beta cells remain to be elucidated.
Observations in human beta cells
Compared to other pathogenic mechanisms of diabetes, dysregulated autophagy is relatively new [18], and research on this topic using human cells is limited (Fig. 2).
T1DM-associated autophagy changes
The role of beta-cell autophagy in T1DM is not well understood [65]. In a recent study, impairment of autophagy was observed in beta cells from organ donors with T1DM: colocalization of lysosomal-associated membrane protein 2 (LAMP-2) with microtubule-associated protein 1 light chain 3 (LC3) (an autophagosome marker) and with proinsulin was significantly reduced in remnant beta cells of individuals with T1DM compared to that in beta cells from non-diabetic individuals; accumulation of telolysosomes with nitrogen-dense rings was observed in beta cells of autoantibody-positive donors, demonstrating that altered lysosomal content occurs before clinical hyperglycemia [66].
T2DM-associated autophagy changes
Signs of dysregulated autophagy were more frequent in beta cells from organ donors with T2DM than in those from non-diabetic donors [67], including increases in the density volume of autophagic vacuoles and autophagosomes, which could be due to either enhanced autophagic activity or blockade of autophagic flux at the lysosomal step [68]. Favoring the latter interpretation, LAMP-2 and cathepsin expression, working at the later stage of autophagy, were decreased in islets from donors with T2DM, along with increased beta cell death [67]. Inhibition of nuclear translocation of transcription factor EB (TFEB), a master regulator of autophagy, and an increase in P62 accumulation, an indicator of reduced autophagic clearance, were also associated with diabetes [69,70]. Taken together, these data indicate that autophagy fails to cope with metabolic challenges in T2DM [13].
Glucolipotoxicity-induced autophagy changes in vitro
Dysregulated autophagy in diabetic beta cells was induced by exposing human islets to FFAs, suggesting that inhibition of autophagic turnover by FFAs is a mediator of lipo- and glucolipotoxicity [67,71]. Similar to rodent cells, a short exposure to saturated fatty acids increased autophagy in human islets, whereas prolonged exposure led to decreased autophagy. However, an even longer exposure was required to compromise autophagy in human islets, with increased LC3-II signals, suggesting that autophagosome formation occurs after up to 48 hours of palmitate exposure [47]. The proposed mechanism of compensatory stimulation of autophagy involves complement C3, which is highly expressed in human pancreatic islets and is correlated with T2DM status, HbA1c, and inflammation [72].
Dysregulated autophagy in beta cells as a therapeutic target
Some chemicals have been shown to regulate autophagy in cells of human origin. In islets from non-diabetic individuals exposed to FFAs and from patients with T2DM, metformin ameliorated autophagy alterations and normalized LAMP-2 expression [67]. Rapamycin, which stimulates autophagy via mTOR complex I (mTORC1), increased insulin secretion and decreased apoptosis [71,73]. However, rapamycin as an immunosuppressive agent induced diabetes in transplant patients. One possible explanation is that rapamycin might deplete ER calcium and reduce mitochondrial calcium uptake in beta cells, leading to decreased insulin release [74]. However, it is not clear whether this is a class effect of mTORC1 inhibitors.
The autophagy enhancer MSL-7 activates calcineurin and nuclear translocation of TFEB without inhibiting mTORC1. MSL-7 reduced hIAPP oligomer accumulation in both human iPS-derived beta cells and 1.1B4 human beta cells incubated with the autophagy inhibitor 3-methyladenine. These findings were dependent on TFEB and were associated with diminished beta cell apoptosis [61].
As mentioned in Section 1.2, the PERK inhibitors GSK260-6414 and GSK2656157 caused islet damage and hyperglycemia in mice [21,22] and significantly downregulated eIF2A phosphorylation. Interestingly, at very low doses that did not significantly affect eIF2A phosphorylation, these inhibitors improved GSIS and hyperglycemia in obese diabetic mice [75]. Recently, we observed that low-dose PERK inhibitors restored autophagy activity in human islets, which was impaired by high glucose and palmitic acid. This restoration was associated with increased insulin content [76]. Therefore, low-dose PERK inhibitors seem to restore human beta cell function under glucolipotoxicity, not via the UPR, but via the autophagy pathway. Our results were obtained using human islets from living donors who underwent partial pancreatectomy for benign diseases, such as cystic adenomas and low-grade neuroendocrine tumors. Pancreatic malignancies in the conditions tumor microenvironment, such as inflammation, might influence beta cell response to the PERK inhibitors [77]. Compared to islets isolated from deceased donors, fresh pancreatic tissues from living donors might better represent physiologic islet cells as the metabolic changes associated with brain death would be minimized, such as fluctuations in glucose metabolism due to medical treatment in the intensive care unit prior to organ donation and autolysis of pancreatic tissues after death [78].
CONCLUSIONS
Human beta cell lines and in vitro differentiated beta cell-like cells do not accurately represent human beta cells. Deceased donor islets might behave differently from living donor islets, but sources of living donor islets are extremely limited. In vivo studies in human cells are challenging, although a model using human islets transplanted into immune-deficient mice has been suggested as a possible approach. However, access to human beta cells for experimental systems is improving, which will facilitate development of novel therapies for diabetes.
If pharmacological modulation of UPR pathways and autophagy in human beta cells proves effective for treating diabetes, the remaining obstacles of bioavailability, solubility, and safety will need to be addressed. These issues could be overcome by targeted delivery using beta cell-specific markers; dosage modification, like our findings with PERK inhibitors; and repurposed drugs, like imatinib.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
This was supported by NRF grants (2019R1A2C1007397 and 2022R1A2C2004570) funded by the Ministry of Science and ICT, Republic of Korea.
ACKNOWLEDGMENTS
None