Exercise, Mitohormesis, and Mitochondrial ORF of the 12S rRNA Type-C (MOTS-c)
Article information

Abstract
Low levels of mitochondrial stress are beneficial for organismal health and survival through a process known as mitohormesis. Mitohormetic responses occur during or after exercise and may mediate some salutary effects of exercise on metabolism. Exercise-related mitohormesis involves reactive oxygen species production, mitochondrial unfolded protein response (UPRmt), and release of mitochondria-derived peptides (MDPs). MDPs are a group of small peptides encoded by mitochondrial DNA with beneficial metabolic effects. Among MDPs, mitochondrial ORF of the 12S rRNA type-c (MOTS-c) is the most associated with exercise. MOTS-c expression levels increase in skeletal muscles, systemic circulation, and the hypothalamus upon exercise. Systemic MOTS-c administration increases exercise performance by boosting skeletal muscle stress responses and by enhancing metabolic adaptation to exercise. Exogenous MOTS-c also stimulates thermogenesis in subcutaneous white adipose tissues, thereby enhancing energy expenditure and contributing to the anti-obesity effects of exercise training. This review briefly summarizes the mitohormetic mechanisms of exercise with an emphasis on MOTS-c.
The Sulwon Award for Scientific Achievement is the Korean Diabetes Association’s highest scientific award and honors an individual who has excellently contributed to the progress in the field of diabetes and metabolism. The Sulwon Award is named after an emeritus professor, Eung Jin Kim, who founded Korean Diabetes Association. Prof. Min-Seon Kim received the 13th Sulwon Award at the 34th Spring Congress of Korean Diabetes Association & 5th Korea-Japan Diabetes Forum which was held as a virtual congress from May 6 to 8 in 2021.
INTRODUCTION
In 2016, the World Health Organization (WHO) reported that 39% of adults aged ≥18 years were overweight, and 13% were obese [1]. Additionally, this report indicated that over 340 million children and adolescents aged 5 to 19 were overweight or obese [1]. Since 1975, the prevalence of overweight and obesity in adults and children has continued to grow every year [1]. A rapid expansion in the overweight or obese population concomitantly increases the prevalence of obesity-associated disorders such as type 2 diabetes mellitus, hypertension, dyslipidemia, heart disease, and certain types of cancer [2,3]. Notably, obesity and overweight are preventable and can be alleviated by increasing physical activity [4]. Exercise reduces excessive fat mass and ultimately helps to maintain a non-obese healthy condition [4]. Exercise provides metabolic advantages by increasing oxygen consumption, insulin sensitivity, and fatty acid oxidation [5]. These beneficial effects of exercise are strongly associated with mitochondrial adaptive changes [6,7]. In this review, we will outline the latest knowledge on the role of mitochondria and the mitochondria-derived peptide (MDP) mitochondrial ORF of the 12S rRNA type-c (MOTS-c) in exercise physiology.
MITOCHONDRIAL ADAPTATION TO EXERCISE
Exercise induces a variety of changes in mitochondria depending on exercise intensity, duration, and frequency [8,9]. These include changes in mitochondrial biogenesis, mitochondrial dynamics (fusion/fission), cristae remodeling, mitophagy, mitochondrial respiration capacity, and oxidative metabolism (Fig. 1). Exercise offers metabolic advantages such as increased oxygen consumption, insulin sensitivity, and fatty acid oxidation, which are closely linked to mitochondrial functions [5,10].
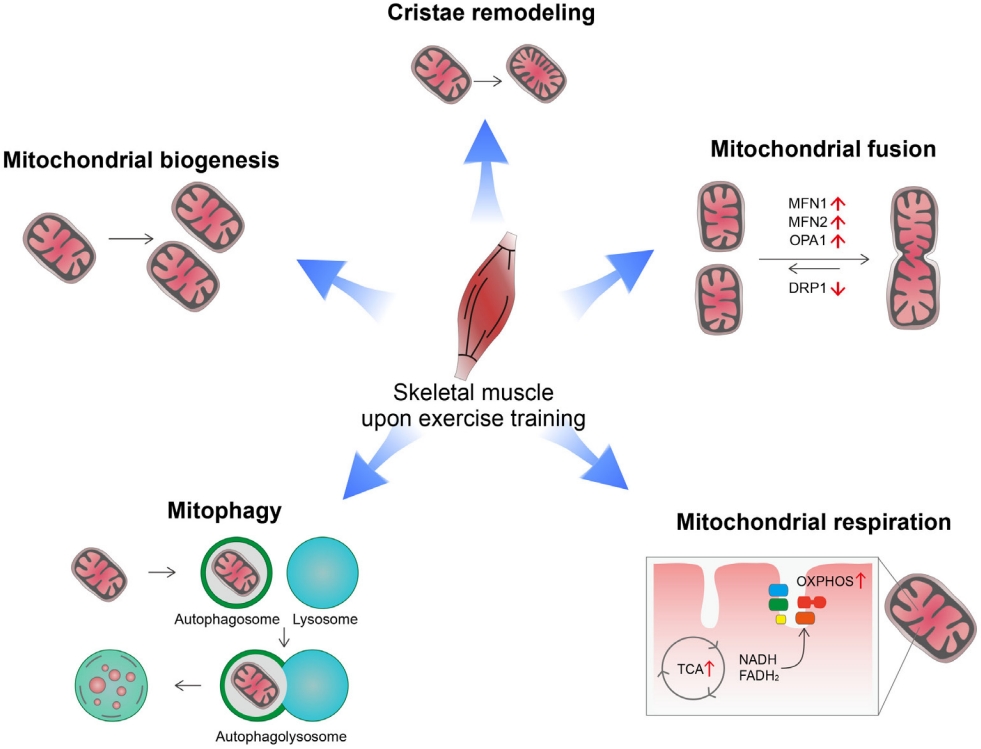
Mitochondrial changes in response to acute exercise or exercise training. Exercise induces a variety of changes in mitochondria depending on the exercise intensity, duration, and frequency. Exercise increases mitochondrial mass through increasing mitochondrial biogenesis and increases inner mitochondrial membrane surface through cristae remodeling. These changes lead to increased mitochondrial respiration and oxidative metabolism. Exercise also promotes overall mitochondrial fusion and autophagy, which may help to maintain mitochondrial function and homeostasis during exercise-induced stress. MFN1, mitofusin 1; MFN2, mitofusin 2; OPA1, optic atrophy 1; DRP1, dynamin related protein 1; OXPHOS, oxidative phosphorylation; TCA: tricarboxylic acid; NADH, nicotinamide adenine dinucleotide (reduced); FADH2, flavin adenine dinucleotide (reduced).
High-intensity physical training enhances mitochondrial biogenesis and improves mitochondrial functional capacity [11]. Mitochondrial biogenesis is a process that synthesizes new mitochondria and is primarily regulated by the peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1-alpha (PGC-1α) [12]. All three isoforms of PGC-1α are involved in skeletal muscle mitochondrial biogenesis [13,14]. Indeed, PGC-1α overexpression increases mitochondrial biogenesis and capillary density in skeletal muscle, which leads to increased mitochondrial catabolic metabolism and enhanced physical activity [13]. Exercise increases PGC-1α expression via the β2-adrenergic receptor and its downstream signaling pathways [15]. Additionally, intracellular Ca2+ levels increase in contracting muscles and act as a messenger for the transcriptional regulation of exercise-related genes. The calcium/calmodulin-dependent protein kinase (CaMK) largely mediates the regulatory functions of Ca2+ [16]. Physical exercise activates CaMK-II, the dominant isoform of CaMK in human skeletal muscle [17]. In turn, CaMK-II stimulates mitochondrial biogenesis and improves glucose transport by stimulating PGC-1α expression [18-20]. These findings indicate an involvement of CaMK-II in exercise-induced mitochondrial and metabolic adaptation.
In addition to mitochondria biogenesis, endurance exercise increases the area of mitochondrial inner membranes per mitochondrial volume in human muscles [21]. Moreover, the mitochondrial cristae density correlates with whole body oxygen uptake and serves as a marker for mitochondrial respiratory capacity [21]. This mitochondrial cristae remodeling may be a part of mitochondrial adaptation for increasing mitochondrial respiration.
Mitochondrial dynamics (i.e., fission and fusion) play critical roles in maintaining mitochondrial functions and morphology [22]. Key regulators of these processes are the cytosolic GTPase dynamin-related protein 1 (DRP1) for fission and mitofusin 1 (MFN1), MFN2, and optic atrophy 1 (OPA1) for fusion [23-25]. Exercise stimulates mitochondrial fusion by increasing the expression of mitofusins (MFN1, MFN2) in human skeletal muscles [26]. Exercise-induced changes in mitochondrial dynamics are mediated via both the PGC-1α and estrogen-related receptor α (ERRα) [26]. Moreover, inhibiting mitochondrial fusion in skeletal muscles via depletion of MFN1/MFN2 impedes exercise performance and shortens exhaustion time in mice [27]. Taken altogether, mitochondrial fusion may be critical for skeletal muscle adaptation to exercise and exercise performance.
Autophagy is a catabolic pathway that recycles intracellular components under energy-shortage conditions and clears damaged organs and protein aggregates [28]. Exercise increases mitochondrial autophagy, i.e., mitophagy in skeletal muscle and heart tissue through AMP activated protein kinase (AMPK)- dependent mechanisms [29]. Exercise-induced mitophagy helps to maintain mitochondrial homeostasis by selective removal of damaged/dysfunctional mitochondria [29]. Therefore, mitophagy is considered an important part of mitochondrial adaptation to exercise.
POTENTIAL MECHANISMS OF EXERCISE-INDUCED MITOHORMESIS
Hormesis refers to salutary biological adaptations to low continuous or moderate intermittent doses of stress, which may be fatal at higher or chronic doses. Exposure to lower levels of stress protects organisms against subsequent greater stress. Mitohormesis, a compound word derived from mitochondria and hormesis, was initially demonstrated in lower organisms such as Caenorhabditis elegans [30,31]. Exposure to low levels of mitochondrial stressors extends life span and retards aging-related diseases [30,31]. Now, this phenomenon has been observed in mammals [32].
Exercise may be a powerful way to induce mitohormesis [33]. Potential mechanisms underlying exercise-induced mitohormesis are depicted in Fig. 2. During exercise or muscle contraction, reactive oxygen species (ROS) are produced in skeletal muscle as a byproduct of mitochondrial oxidative phosphorylation (OXPHOS) [33]. Chronic ROS overproduction by excessive metabolic flux and mitochondrial dysfunction, have been implicated in the progression of human diseases such as atherosclerosis, diabetes, dementia, and cancer [34-36]. In contrast, moderate levels of ROS, such as is generated during exercise, may help maintain normal energy metabolism and health [33]. Exercise-induced ROS production increases the expression of PGC-1α, nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (mtTFA)/mitochondrial transcription factor A (TFAM) in skeletal muscle [33]. Moreover, supplementation with antioxidants attenuates exercise-induced PGC-1α expression, mitochondrial biogenesis, and insulin sensitivity in skeletal muscles [37]. In line with this, a chronic moderate degree of exercise training increases ROS production in the hypothalamus, a controlling center of energy balance [38]. Hypothalamic ROS production during exercise training is critical for exercise-induced thermogenesis given that central administration of antioxidants blocks exercise-induced thermogenesis [38].

Mitohormetic responses to exercise. Exercise induces beneficial stress responses in mitochondria in a process known as mitohormesis. Exercise increases the production of reactive oxygen species (ROS) and triggers mitochondrial unfolded protein responses (UPRmt). However, exercise-induced mitochondrial stress also stimulates the production and release of myomitokines (growth differentiation factor 15 [GDF15] and fibroblast growth factor 21 [FGF21]) as well as mitochondrial DNA (mtDNA)-derived peptides (humanin and mitochondrial ORF of the 12S rRNA type-c [MOTS-c]), all of which have beneficial metabolic effects. PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; NRF1, nuclear respiratory factor 1; mtTFA, mitochondrial transcription factor A; JNK, c-Jun N-terminal kinase; eIF2α, eukaryotic initiation factor-2α; ATF4, activating transcription factor 4; AP-1, activating protein-1; CHOP, C/EBP-homologous protein; eIF1α-P, phosphorylated eukaryotic initiation factor-1α; ISR, integrated stress response.
The mitochondrial unfolded protein response (UPRmt) is a type of adaptive response to mitochondrial stress that occurs during recovery from various mitochondrial insults [39-41]. Upon mitochondrial stress, unfolded or misfolded proteins accumulate in the mitochondria and are subsequently degraded by mitochondrial proteases and exported to the cytosol [39, 41]. These proteins activate retrograde signaling involving activating transcription factor associated with stress-1 (ATFS-1) in C. elegans and c-Jun N-terminal kinase (JNK) and activating transcription factor 4 (ATF4) in mammals and relay mitochondrial stress signals to the nucleus [39,41]. Consequently, the transcription of mitochondrial chaperones and proteases is stimulated to help resolve mitochondrial proteotoxic stress. Evidence suggests that induction of UPRmt improves survival and provides a health benefit [39-41]. Interestingly, UPRmt also takes place in neighboring cells or distal tissues through a process known as non-cell-autonomous UPRmt [42]. For instance, knocking down electron transport chain components in the nervous system can trigger UPRmt responses in the intestine and extend the life span of C. elegans [42]. It was recently shown that high-intensity interval training induces UPRmt in the skeletal muscle of aged mice [43]. Moreover, we observed that moderate-intensity treadmill exercise induced UPRmt in the white and brown adipose tissue (BAT) of young mice [38]. These data suggest that UPRmt may underlie the mitohormetic effect of exercise.
Exercise-induced mitohormetic responses may be also mediated by myomitokines such as fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15) [44,45]. The expression levels of both factors in skeletal muscle are elevated by exercise-induced mitochondrial stress via the integrated stress response (ISR) involving ATF4-C/EBP-homologous protein (CHOP) pathway [44,45]. They are released into the bloodstream to stimulate adipose tissue thermogenesis, lipolysis, and fatty acid oxidation, leading to alleviation of obesity and obesity-related metabolic complications [46-48].
Another potential endocrine mediator of exercise-induced mitohormesis is mitochondrial DNA (mtDNA)-encoded small proteins, called MDPs [49], given that their production is increased by exercise and mitochondrial stress and depends on ROS levels [38]. The roles of MDPs in exercise physiology will be described in detail in the following section.
MITOCHONDRIA-DERIVED PEPTIDES AND THEIR BIOLOGICAL ACTIONS
MDPs are small bioactive peptides encoded by short open-reading frames (sORF) in mtDNA. To date, eight MDPs have been identified, most of which have various cell- and organ-protective properties [38,50]. Humanin was the first MDP to be identified and is encoded by an sORF in the 16S ribosomal RNA gene called MT-RNR2 [51,52]. Humanin has been identified as a cytoprotective and anti-apoptotic factor in Alzheimer’s diseases, where it protects neurons from amyloid-β-related cytotoxicity [52,53]. Its neuroprotective effects are mediated via inhibition of JNK, activation of signal transducer and activator of transcription-3 (STAT3) and extracellular signal-regulated kinase (ERK) signaling as well as through interactions with insulin-like growth factor-binding protein 3 (IGFBP-3) [54,55]. Humanin also has beneficial roles in glucose metabolism. Indeed, humanin treatment improves glucose-stimulated insulin secretion and peripheral insulin sensitivity [56,57]. Another six small humanin‐like peptides (SHLP) are encoded in the ORFs within the same 16S rRNA gene in which humanin is located [58]. SHLP-2 and SHLP-3 have similar protective effects as humanin and improve mitochondrial metabolism by increasing oxygen consumption rate and reducing apoptosis and ROS production [58]. SHLP-2 and SHLP-3 act as insulin sensitizers [58]. Central and systemic treatment with SHLP-2 and SHLP-3 enhance the ability of insulin to inhibit hepatic glucose production and to stimulate glucose uptake [58]. Both peptides also promote adipocyte differentiation in 3T3‐L1 preadipocytes [58].
A novel MDP named MOTS-c was recently identified [59]. Unbiased gene microarray and global metabolomics assays revealed that the folate-methionine cycle is a target of MOTS-c function [59]. By inhibiting the folate cycle at the level of 5-methyl-tetrahydrofolate (5Me-THF), MOTS-c treatment for 4 hours increases the cellular levels of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) in cultured cells [59]. Resultant AMPK activation improves glucose and fatty acid metabolism. Seven days MOTS‐c infusion significantly increases glucose clearance and the insulin‐stimulated glucose disposal rate in lean mice [59]. This effect may be mediated via through increased glucose transporter 4 (GLUT4) expression and mitochondrial fusion-induced GLUT4 translocation to the plasma membrane [59,60].
Moreover, chronic systemic administration of MOTS‐c for 8 weeks prevents high-fat diet (HFD)‐induced obesity and insulin resistance [59]. MOTS-c treatment also improved hyperglycemia and reproductive outcomes in the mice model of gestational diabetes mellitus [61] and attenuated the development of autoimmune diabetes by suppressing T-cells-induced β-cell destruction in mice [62]. Subsequent studies have demonstrated multiple beneficial effects of MOTS-c on bone biology, inflammation, pain, vascular calcification, and myocardial remodeling where these actions seem to be largely mediated via AMPK activation [63-66].
EFFECTS OF EXERCISE ON THE EXPRESSION OF MDPs
Exercise-induced changes in MDP expression have been investigated in humans and mice [67-69]. In humans, acute high-intensity endurance exercise (cycling) increased plasma concentrations of humanin, SHLP-6, and MOTS-c, as well as the skeletal muscle expression of humanin and MOTS-c [67-69]. Acute exercise-induced elevations in blood and muscle MOTS-c expression returned to baseline levels several hours later [68]. Exercise-induced changes in MDPs may differ according to exercise types. Von Walden et al. [69] showed that plasma humanin levels were elevated during post-endurance exercise (cycling) but remained unaltered after resistance exercise (leg press and extension). The effects of exercise training on MDP expression are less distinct than those of acute exercise [67,70,71]. Twelve weeks of endurance exercise (Nordic walk) in older men with prediabetes increased humanin protein levels in the serum but not in skeletal muscle [70]. Sixteen weeks of combined endurance and resistance exercise increased MOTS-c levels in the blood of non-Hispanic breast cancer survivors [71]. In contrast, 2 weeks of high-intensity cycling in young men and 8 weeks of endurance exercise in women with polycystic ovary syndrome did not alter the expression levels of humanin, SHLP-2, or MOTS-c in the blood and skeletal muscles [72]. In mice, eight weeks of treadmill running in lean and obese male mice elevated the plasma and skeletal muscle expression levels of MOTS-c [73,74]. We found that in young male mice, a moderate-degree of treadmill exercise for 2 weeks increased MOTS-c expression in the hypothalamus while acute running until exhaustion had no effect [38].
It remains unclear how exercise alters MDP expression. Upon acute exercise, humanin protein expression in skeletal muscles increases within 30 minutes without changes in its transcript levels [67]. Moreover, humanin directly binds to E3 ubiquitin-protein ligase tripartite motif 11 (TRIM11), implying the possibility of proteolysis-mediated regulation of humanin [75]. This regulatory mechanism has not been reported for MOTS-c. The expression of MDPs may be also regulated at the transcriptional level. Indeed, adiponectin increases MOTS-c expression in a sirtuin 1 (SIRT1)- and PGC-1α-dependent manner [74], suggesting the involvement of PGC-1α in the transcriptional regulation of MOTS-c.
EFFECTS OF MOTS-c TREATMENT ON EXERCISE PERFORMANCE
Among MDPs, MOTS-c is the most associated with exercise. While exercise controls MOTS-c expression, MOTS-c controls exercise performance. A recent paper reported that chronic MOTS-c treatment enhanced physical activity and health span in young-, middle-, and old-age mice [68]. Mice treated with MOTS-c for 2 weeks displayed increased muscle force and stride length, along with increased lean mass and decreased fat mass [68]. However, the detailed mechanisms by which MOTS-c increases physical performance largely remain to be elucidated. Notably, it took more than 1 week to observe the effect of MOTS-c on exercise performance [68]. Thus, MOTS-c may promote adaptive responses to exercise-induced metabolic and oxidative stress in skeletal muscle (Fig. 3A). Supporting this assumption, MOTS-c treated mice exhibit increased expression of genes associated with glucose and amino acid metabolism and metabolic stress responses in skeletal muscles [68]. Particularly, the expression of heat shock factor 1 (HSF1), heme oxygenase-1 (HO-1), nuclear factor erythroid-2-related factor 2 (NFE2L2/NRF2) is significantly increased [68] and these genes are linked to exercise performance enhancement [76-78]. HSF1 is associated with maintaining protein quality and metabolism, especially under stress conditions [79]. HO-1 and NRF2 are associated with antioxidant capacity and maintenance of mitochondrial function [76-78]. Therefore, these molecules may act downstream of MOTS-c to affect physical performance. As shown previously [80], intracellular MOTS-c might translocate to the nucleus during exercise, where it may possibly modulate the transcription of those genes by interacting with transcription factors and coregulators. Additionally, MOTS-c treatment increases the expression of mitochondrial biogenesis-related molecules such as PGC-1α, NRF1, TFAM, cytochrome c oxidase subunit 4 (COX4), and translocase of outer mitochondrial membrane 20 (TOMM20) and induces mitochondrial fusion by upregulating fusion-related genes such as MFN2 and OPA1 [60]. These mitochondrial changes can also contribute to MOTS-c-enhanced physical performance. In addition, a recent paper has shown that MOTS-c treatment for 8 weeks prevented HFD-induced muscle atrophy through suppression of myostatin expression [81].

Roles of the mitochondria-derived peptide mitochondrial ORF of the 12S rRNA type-c (MOTS-c) in exercise physiology. (A) MOTS-c production is increased in exercising skeletal muscle in a reactive oxygen species (ROS)-dependent manner. MOTSc translocates to the nucleus to modulate gene expression profiles involved in stress adaptation, mitochondrial biogenesis, and mitochondrial dynamics. This mechanism may contribute to enhanced exercise capacity induced by exercise training. (B) Exercise enhances MOTS-c expression in hypothalamic proopiomelanocortin (POMC) neurons, likely through exercise-related myokines, such as interleukin-6 (IL-6). MOTS-c stimulates POMC transcription and β-endorphin production, which in turn increases sympathetic nerve activity innervating the inguinal subcutaneous white adipose tissue, and consequently, the beiging of inguinal subcutaneous white adipose tissue (iWAT) and enhanced thermogenesis are induced. This mechanism may underlie exerciseinduced thermogenesis. AMPK, AMP activated protein kinase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; HSF1, heat shock factor 1; HO-1, heme oxygenase-1; NRF2, nuclear respiratory factor 2; ARH, arcuate nucleus of the hypothalamus; 3V, the third cerebroventricle; SNS, sympathetic nervous system.
MOTS-c is secreted from exercising muscles [68,74], and extracellular MOTS-c may act on neighboring cells or cells in remote organs like hormones [82]. It is yet unclear whether intracellular and extracellular MOTS-c are structurally different. Indeed, MOTS-c treatment stimulates fatty acid oxidation and glucose uptake in skeletal muscle cells and adipocytes through increased AMPK activity [59,83]. At the same time, MOTS-c may interact with other metabolic regulators. For example, adiponectin stimulates MOTS-c expression and vice versa [74]. These two molecules act synergistically to stimulate adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1), SIRT1, and PGC-1α expression [74]. A recent study has demonstrated that exercise training and MOTS-c treatment have additive effects on weight loss, improvement of insulin resistance and PGC-1α upregulation [73]. Thus, MOTS-c and exercise training may have some non-overlapping effects.
MOTS-c AND EXERCISE-INDUCED THERMOGENESIS
In addition to skeletal muscle, exercise training causes beneficial adaptations to multiple organs, including adipose tissues [84,85]. The most prominent finding of exercise-induced changes in adipose tissue is ‘beiging’ or ‘browning’ in the inguinal subcutaneous white adipose tissue (iWAT) [84,85]. Adipocytes in this fat depot look like classical white adipocytes under the conditions of low thermogenic need. However, upon increased thermogenic needs induced by cold exposure, HFD feeding, and exercise, these cells adopt the features of brown adipocytes, i.e., small multilocular fat droplets, high mitochondrial density/activity, increased glucose uptake, and increased expression of thermogenic genes such as uncoupling protein-1 (UCP1), PR domain containing 16 (PRDM16), and PGC-1α. Exercise-induced changes are relatively less distinct in visceral WAT and the primary thermogenic organ BAT [85]. Therefore, exercise-induced adipose tissue thermogenesis seems to occur primarily in the iWAT. On the other hand, skeletal muscle non-shivering thermogenesis also increases during physical activity, possibly through sarcoplasmic reticulum calcium ATPase (SERCA)-dependent mechanisms [86]. It is thus curious why adipose tissue thermogenesis should be elevated despite increased skeletal muscle thermogenesis.
The major mediator of exercise-induced thermogenesis is thought to be the sympathetic nerves innervating iWAT [87]. Other potential mediators are exercise-induced myokines, such as irisin [88], myostatin [89], meteorin-like 1 (Metrnl) [90], lactate [91], and β-aminoisobutyric acid (BAIBA) [92]. MOTS-s can also be considered a myokine involved in exercise-induced thermogenesis. Indeed, intraperitoneal injection of MOTS-c for 7 days promotes cold tolerance by stimulating non-shivering thermogenesis [93]. Mechanistically, it is thought that MOTS-c dramatically upregulates the thermogenic gene expression in BAT and induces beiging in the iWAT [93]. Interestingly, MOTS-c can stimulate thermogenic activity through central mechanisms (Fig. 3B). In our previous study [38], moderate-degree exercise training increases MOTS-c expression in hypothalamic neurons via exercise-related myokine interleukin-6. Moreover, intracerebroventricular administration of MOTS-c for 28 days induced beiging of iWAT and enhanced thermogenic gene expression [38]. These effects occurred via increased β-endorphin production in hypothalamic proopiomelanocortin (POMC) neurons and enhanced sympathetic innervation in iWAT [38]. In this process, hypothalamic neuron ROS production may be essential given that inhibition of hypothalamic ROS production during exercise training significantly inhibits the inducible thermogenic response in iWAT [38].
CONCLUSIONS
Exercise is a type of stress to our body as it depletes energy stores, stimulates ROS production and induces mitochondrial stress responses, such as UPRmt. Mitochondrial stress responses to exercise generally yield beneficial outcomes by increasing mitochondrial anti-oxidant and protein-folding capacities [33]. These reactions are currently proven in skeletal muscles, adipose tissues, and the hypothalamus [38,43,94,95] and might also occur in other organs. Future studies are warranted to test this possibility.
MDPs have drawn attention from many researchers as this class of peptides has numerous beneficial biological effects on the brain, cardiovascular system, glucose/energy metabolism, and bones [54,59,70,71]. Moreover, MDPs are short peptides consisting of less than 30 amino acids [50], potentially making them more therapeutically approachable. In this view, MOTS-c is a promising target as an exercise mimetic or physical performance enhancer. Indeed, MOTS-c treatment enhances physical capacity and metabolic health in both young and aged mice [68]. This effect must be confirmed in humans, especially in physically inactive elderly persons. In addition, as both MOTS-c and antidiabetic drug metformin activate AMPK [59, 96], it will be worth to test whether MOTS-c/metformin combination treatment will be more effective than monotherapy in activating AMPK and improving glucose metabolism. To be druggable, MOTS-c might be modified to degradation-resistant form for long-lasting effects. Better insight into the adaptive responses to exercise can lead us to challenging intervention for many human diseases, including obesity.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
This study was supported by grants from the National Research Foundation of Korea (NRF), the Ministry of Education of Korea (2020R1A2C3004843, 2020R1A4A3078962, 2022R1C1C1004187), and the Asan Institute for Life Sciences (2019-IP0855-1).
Acknowledgements
We thank the Scientific Publications Team at Asan Medical Center.