Treatment of Diabetic Kidney Disease: Current and Future
Article information

Abstract
Diabetic kidney disease (DKD) is the major cause of end-stage kidney disease. However, only renin-angiotensin system inhibitor with multidisciplinary treatments is effective for DKD. In 2019, sodium-glucose cotransporter 2 (SGLT2) inhibitor showed efficacy against DKD in Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) trial, adding a new treatment option. However, the progression of DKD has not been completely controlled. The patients with transient exposure to hyperglycemia develop diabetic complications, including DKD, even after normalization of their blood glucose. Temporary hyperglycemia causes advanced glycation end product (AGE) accumulations and epigenetic changes as metabolic memory. The drugs that improve metabolic memory are awaited, and AGE inhibitors and histone modification inhibitors are the focus of clinical and basic research. In addition, incretin-related drugs showed a renoprotective ability in many clinical trials, and these trials with renal outcome as their primary endpoint are currently ongoing. Hypoxia-inducible factor prolyl hydroxylase inhibitors recently approved for renal anemia may be renoprotective since they improve tubulointerstitial hypoxia. Furthermore, NF-E2–related factor 2 activators improved the glomerular filtration rate of DKD patients in Bardoxolone Methyl Treatment: Renal Function in chronic kidney disease/Type 2 Diabetes (BEAM) trial and Phase II Study of Bardoxolone Methyl in Patients with Chronic Kidney Disease and Type 2 Diabetes (TSUBAKI) trial. Thus, following SGLT2 inhibitor, numerous novel drugs could be utilized in treating DKD. Future studies are expected to provide new insights.
INTRODUCTION
Diabetic kidney disease (DKD) is one of the major complications for diabetic patients and the most significant cause of end-stage kidney disease (ESKD) [1]. Until now, chronic kidney disease (CKD) caused by diabetes mellitus is diagnosed as diabetic nephropathy, which begins with microalbuminuria, followed by macroalbuminuria and then gradual decline in kidney function, and is diagnosed pathologically by characteristic pathological findings, such as increased mesangial substrate, nodular lesions, and tubulointerstitial fibrosis [2]. However, in recent years, cases of impaired renal function without albuminuria have been reported [3,4]. In this background, a new disease concept called DKD was born. DKD is defined as CKD with diabetes being partially involved in the pathogenesis of kidney disease, encompassing the concept of classical diabetic nephropathy (Fig. 1) [5].

The concept of diabetic kidney disease and diabetic nephropathy. GFR, glomerular filtration rate.
The pathogenesis of DKD includes glomerular hypertension, change of renal hemodynamics, ischemia and hypoxia, oxidative stress, and upregulation of the renin-aldosterone system [6]. However, the full pathogenesis of the disease remains to be understood, and therapeutic targets have not been determined. Therefore, the treatment strategy for DKD remains the same with that of conventional diabetic nephropathy. Multi-disciplinary treatments, including blood glucose control, blood pressure and lipid control with renin-angiotensin-aldosterone system (RAS) inhibitors, appropriate weight management, and guidance for diet and smoking cessation, are important. Aside from multidisciplinary treatments, sodium-glucose cotransporter 2 (SGLT2) inhibitors were also added as a new drug of choice for DKD treatment in 2019, as the Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) study showed that SGLT2 inhibitors inhibited DKD progression [7]. However, even with the use of RAS and SGLT2 inhibitors, 5.27% (116/2,202) of patients continue to develop ESKD in the CREDENCE study.
One of the important factors in DKD development is the phenomenon known as “metabolic memory.” Patients with past exposure to hyperglycemia have been known to develop complications, including DKD, even after treatment has normalized their blood glucose levels [8,9]. Past hyperglycemia is considered to have led to the accumulation of advanced glycation end product (AGE) and epigenetic genetic changes, such as DNA methylation and histone modifications, which may have led to the progression of DKD as a cellular memory [10,11]. These DKD mechanisms have the potential as novel therapeutics for DKD. This paper outlines current and future therapies for DKD.
CURRENT TREATMENT
RAS inhibitor
Angiotensin-converting-enzyme inhibitor and angiotensin II receptor blocker
The oldest and most preeminent drug for DKD treatment is the RAS inhibitor. Actually, RAS inhibitors have been shown to be effective in treating DKD in many clinical trials. In 1993, the Captopril Collaborative Study published by Lewis et al. [12] showed that captopril, angiotensin-converting-enzyme inhibitor (ACE-I), inhibited the progression of nephropathy in patients with overt nephropathy in type 1 diabetes mellitus (Fig. 2). In addition, angiotensin II receptor blocker (ARB) in diabetic patients with manifest nephropathy has been demonstrated in large randomized controlled trials (RCTs), such as the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) and Irbesartan in Diabetic nephropathy Trial (IDNT) [13,14]. Moreover, a number of reports on microalbuminuria among DKD patients have been noted. In the Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study (IRMA-2), irbesartan was reported to inhibit the progression from microalbuminuria to overt proteinuria by approximately 70% [15]. The Incident to overt: Angiotensin II receptor blocker, Telmisartan, Investigation On type II diabetic Nephropathy (INNOVATION) study in Japan also reported telmisartan to inhibit the progression from microalbuminuria to overt proteinuria by approximately 60% compared to placebo [16]. Furthermore, Microalbuminuria reduction with valsartan (MARVAL), which compared an ARB with a calcium channel blocker, showed that only ARB was effective in lowering microalbuminuria, indicating that RAS inhibitors have an inhibitory effect on nephropathy and a blood pressure-lowering ability [17]. Large RCTs, Bergamo Nephrologic Diabetes Complications Trial (BENEDICT) and Randomized Olmesartan and Diabetes Microalbuminuria Prevention (ROADMAP), have also demonstrated that ARBs reduced the incidence of microalbuminuria among patients who did not present with microalbuminuria [18,19]. However, RAS inhibitors have also been reported to not be significantly effective in preventing the development of microalbuminuria among patients who are normotensive and without microalbuminuria [20]. Therefore, RAS inhibitors should be used in patients presenting with either hypertension or nephropathy.
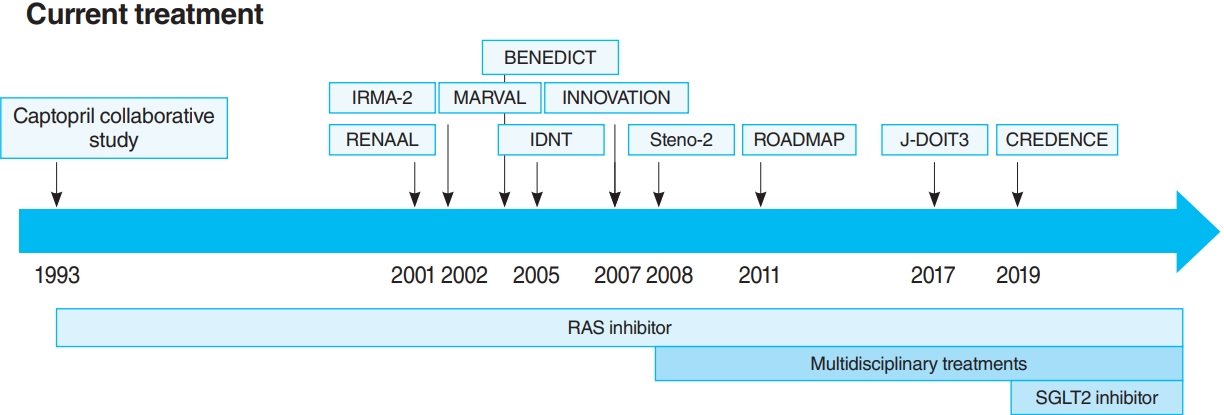
The time course of current treatments. BENEDICT, Bergamo Nephrologic Diabetes Complications Trial; IRMA-2, Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study; MARVAL, Microalbuminuria reduction with valsartan; INNOVATION, Incident to overt: Angiotensin II receptor blocker, Telmisartan, Investigation On type II diabetic Nephropathy; RENAAL, Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan; IDNT, Irbesartan in Diabetic Nephropathy Trial; ROADMAP, Randomized Olmesartan and Diabetes Microalbuminuria Prevention; J-DOIT3, Japan Diabetes Optimal Integrated Treatment study for 3 major risk factors of cardiovascular disease; CREDENCE, Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation; RAS, renin-angiotensin system; SGLT2, sodium-glucose cotransporter 2.
In this way, RAS inhibitors have been shown to be effective in all stages of DKD. The combination therapy of ACE-I and ARB was also studied in large randomized trials. Although some studies have shown combination therapy to be beneficial [21], the Ongoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET) reported a significant increase in worsening kidney function and mortality in the telmisartan-ramipril combination treatment group [22]. Furthermore, in the Veterans Affairs nephropathy in Diabetes (VA Nephron D) study, no significant difference was noted in the efficacy for DKD of the combination treatment group, and the increase in adverse events, such as hyperkalemia and acute kidney injury (AKI), has been reported [23]. Based on these results, the combination of ACE-I and ARB is not recommended.
Overactivation of RAS causes intraglomerular hypertension due to constricted efferent arterioles, leading to increased proteinuria, mesangial cell proliferation, and activation of inflammatory responses and fibrotic factors [24,25]. Apart from the glomerular hypertension mechanism, angiotensin II is also known to cause cellular hypertrophy, accumulation of extracellular matrix and production of reactive oxygen species, and inflammatory changes by the production of cytokines, such as transforming growth factor (TGF)-β in glomerular cells [26,27]. Angiotensin II has also been reported to induce apoptosis of podocytes and decrease tubular interstitial microvessels and chronic hypoxia [28,29]. Moreover, aldosterone has been reported to cause increased expression of plasminogen activator inhibitor-1 (PAI-1) and TGF-β, activation of macrophages, and endothelial cell damage by a different mechanism from glomerular hypertension [30]. RAS inhibitors inhibit DKD progression by suppressing these mechanisms caused by RAS overactivation.
Mineralocorticoid receptor antagonist
Mineralocorticoid receptor antagonist (MRA) has an antihypertensive effect by suppressing the action of aldosterone, the end product of RAS and has been reported to reduce proteinuria. For example, spironolactone was reported to be beneficial in decreasing albuminuria, inflammation, and fibrosis among DKD patients [31], and eplerenone was also reported to reduce albuminuria more than enalapril among systemic hypertension patients [32]. However, these drugs cause hyperkalemia in CKD patients and are contraindicated in patients with impaired renal function. In this context, new MRAs, finerenone and esaxerenone, have been developed, which are more selective for mineralocorticoid receptor, have no steroid structure, and have fewer complications than spironolactone and eplerenone. They have also been reported to reduce proteinuria among DKD patients [33,34]. In Japan, esaxerenone was approved for hypertension in 2019 and, unlike spironolactone and eplerenone, has been used for DKD patients with microalbuminuria and patients with moderate renal impairment. Furthermore, the large phase III studies, Finerenone in reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease (FIDELIO-DKD) and Finerenone in Reducing Cardiovascular Mortality and Morbidity in Diabetic Kidney Disease (FIGARO-DKD), are evaluating, evaluate the efficacy and safety of finerenone in DKD patients, focusing on the composite primary endpoints of renal and cardiovascular functions [35,36].
In this way, both large clinical trials and basic experiments have a solid evidence that RAS inhibitors, especially ACE-I and ARB, can treat DKD. However, although RAS inhibitors reduce proteinuria and slow the decline of glomerular filtration rate (GFR), they cannot suppress ESKD development completely. In addition, they increase the incidence of complications, such as AKI and hyperkalemia. Therefore, a new drug for DKD was expected to come out.
Importance of multidisciplinary treatments
For a long time, there were no drugs specific to DKD other than RAS inhibitors. In this context, some papers reported that multidisciplinary treatment, such as blood glucose control, blood pressure control with RAS inhibitor, lipid control, and lifestyle modifications, significantly reduced cardiovascular events in DKD patients and, as a secondary endpoint, suppressed renal events. The most famous trial that demonstrated the effectiveness of multidisciplinary treatment was the Steno-2 study (Fig. 2) [37]. The Steno-2 trial reported that multidisciplinary treatment not only reduced cardiovascular events among DKD patients but also retarded the progression of DKD itself. However, some problems with the Steno-2 study were noted, including the small number of participants and the much higher mean glycosylated hemoglobin (HbA1c) than the current guideline targets even in the intensive-therapy group (HbA1c, 7.9%). From this background, Japan Diabetes Optimal Integrated Treatment Study for 3 major risk factors of cardiovascular disease (J-DOIT3) study was reported in 2017, which examined the efficacy of multifactor intensification therapy for the prevention of cardiovascular complications among Japanese type 2 diabetes mellitus patients (Fig. 2) [38, 39]. The J-DOIT3 study included 2,542 type 2 diabetes mellitus patients, with a target HbA1c level of 6.2% in the intensive treatment group. Similar to the Steno-2 study, J-DOIT3 showed a nonsignificant reduction in cardiovascular events and an inhibitory effect on DKD progression in the multidisciplinary treatment group.
SGLT2 inhibitor
SGLT2, which is a cotransporter, transports sodium and glucose in 1:1 ratio using Na-K ATPase-mediated sodium concentration gradient on the proximal tubular basement membrane. SGLT2 inhibitors inhibit proximal tubular glucose reabsorption and promote urinary glucose excretion, resulting in hypoglycemia. Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME), published in 2015, showed that empagliflozin significantly reduces cardiovascular events in type 2 diabetes mellitus patients with pre-existing cardiovascular disease [40], and a post hoc analysis of renal outcomes showed that empagliflozin significantly reduced the progression of nephropathy [41]. Aside from empagliflozin, the results of the Canagliflozin Cardiovascular Assessment Study (CANVAS) Program in canagliflozin [42] and the Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARETIMI58) study in dapagliflozin [43], as well as the systematic review and meta-analysis of these three studies, demonstrated similar cardiovascular and kidney protective effects [44]. However, all the large trials validated the primary endpoint to be cardiovascular events and included patients with a low risk for kidney disease, which has resulted in a low number of kidney events. CREDENCE study was conducted in type 2 diabetes mellitus patients who developed overt albuminuria (estimated glomerular filtration rate [eGFR] 30 to 90 mL/min/1.73 m2, albuminuria 300 to 5,000 mg/gCr), the primary outcome of which being kidney disease events (Fig. 2) [7]. CREDENCE trial achieved prespecified efficacy criteria in July 2018, which was stopped early, demonstrating the efficacy of SGTL2 inhibitors for DKD in type 2 diabetes mellitus patients.
The renoprotective effect mechanisms of SGLT2 inhibitor are not fully understood, the most important of which are the correction of glomerular hyperfiltration and improvement of kidney hypoxia. Glomerular hyperfiltration is known to be responsible for the risk of the appearance of new albuminuria and a decrease in eGFR [45]. SGLT2 inhibitors are thought to have a renoprotective effect by increasing Na reaching the macula densa cells of the distal tubules, thereby correcting the dilation of afferent arterioles and glomerular hyperfiltration by the tubuloglomerular feedback (TGF). They are also thought to have the potential in improving hypoxia in the kidney. A rat model of diabetic nephropathy has been reported to show that oxygen consumption in proximal tubular cells was approximately doubled and that treatment with phlorizin, an SGLT1/2 inhibitor, reduced Na-K ATPase activity, inhibited the increase in oxygen consumption, and improved renal cortex oxygenation [46]. SGLT2 inhibitors are also known to cause a mild increase of ketones in the blood. Compared to glucose and fatty acids, ketone bodies can produce more ATP with small amounts of oxygen, contributing to the improvement of kidney hypoxia [47]. Blocking the intracellular influx of glucose in the proximal tubules reduces mitochondrial damage, leading to anti-inflammatory and antifibrosis results via inhibition of oxidative stress [48]. Furthermore, a comprehensive search for glycolytic and tricarboxylic acid (TCA) cycle metabolites in the kidney by imaging mass spectrometry revealed the inhibition by SGLT2 inhibitor treatment of the accumulation of TCA intermediate metabolites and the reduction of oxidative stress in the glomerulus [49].
The advantages of SGLT2 inhibitor for the treatment of DKD include its efficacy when combined with RAS inhibitor and its ability to treat DKD independently of the hypoglycemic effect. Therefore, it is possible that SGLT2 inhibitor may be effective in the treatment of other CKD forms. A Study to Evaluate the Effect of Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients With Chronic Kidney Disease (DAPA-CKD) study, a phase III study of dapagliflozin, reported that dapagliflozin was effective in patients with CKD, with or without type 2 diabetes mellitus [50]. Furthermore, the Study of Heart and Kidney Protection with empagliflozin (EMPA-KIDNEY) study, a phase III study of empagliflozin, are currently underway for the application of SGLT2 inhibitors for CKD other than DKD, the result of which is expected to be published in the near future. However, the combination therapy with RAS and SGLT2 inhibitor is still only effective in slowing the increase in GFR, with a hope for development of new therapies.
FUTURE TREATMENT
NF-E2–related factor 2 activator
Although current treatments, such as RAS inhibitor and SGLT2 inhibitor, can only slow the decline of GFR, NF-E2–related factor 2 (Nrf2) activator is a novel drug with the potential to improve the GFR of DKD patients [51]. Bardoxolone methyl is a drug that activates Nrf2, which is a transcription factor responsible for the defense response to oxidative stress. Under unstressed conditions, Nrf2 is ubiquitinated and degraded by the proteasome through the action of Kelch-like ECH-associated protein 1 (Keap1), whereas under oxidative stress, Keap1 undergoes various chemical modifications, which reduce its affinity for Nrf2 and inhibit its degradation. Nrf2, which has escaped degradation by Keap1, enters the nucleus to form heterodimers with small Maf proteins and binds to antioxidant responsive elements (ARE) for the enhancement of the expression of downstream genes involved in antioxidant and anti-inflammatory activities [52]. Bardoxolone methyl binds to Keap1 and undergoes a conformational change to promote Nrf2 dissociation, allowing Nrf2 to migrate into the nucleus, and exert antioxidant and anti-inflammatory effects [53].
In 2011, the result of 52-Week Bardoxolone Methyl Treatment: Renal Function in chronic kidney disease/Type 2 Diabetes (BEAM) study, a phase II placebo-controlled trial in DKD patients, was published (Fig. 3) [54]. The BEAM study subjects had DKD with type 2 diabetes mellitus and 20 to 45 mL/min/1.73 m2 eGFR and were randomized into the placebo or bardoxolone methyl group. The bardoxolone methyl group showed a significantly greater increase in eGFR than the placebo group, which persisted for up to 52 weeks, making bardoxolone methyl the major focus for DKD treatment. The Bardoxolone Methyl Evaluation in Patients with CKD and Type 2 Diabetes Mellitus: the Occurrence of Renal Events (BEACON) study is a larger phase III placebo-controlled study than BEAM, which was conducted to provide further evidence of the renoprotective effects of bardoxolone methyl (Fig. 3) [55]. The subjects had DKD with an eGFR of 15 to 30 mL/min/1.73 m2. This study included patients with a more advanced renal impairment than those in the BEAM study. These patients were randomized into placebo or bardoxolone methyl (20 mg/day) group. However, the significantly increased rates of heart failure-related hospitalizations and deaths in the bardoxolone methyl group were observed, resulting in early termination of the study (after 9 months of observation). Despite the early termination, a significant increase in the eGFR levels of the bardoxolone methyl group was still noted. In addition, a post hoc analysis of the BEACON study showed two risk factors for developing heart failure: brain natriuretic peptides (BNPs) ≥200 pg/mL and a history of hospitalization due to heart failure [56]. For patients without these two factors, the incidence of heart failure in the bardoxolone methyl and placebo groups was very low (2%). With this background, the Phase II Study of Bardoxolone Methyl in Patients with Chronic Kidney Disease and Type 2 Diabetes (TSUBAKI) trial was conducted in Japan (Fig. 3) [57], which included patients with G3-4 DKD without heart failure risk factors such as BNP ≥200 pg/mL or a history of hospitalization for heart failure. The bardoxolone methyl group had a significant increase in GFR of 6.6 mL/min/1.73 m2 over the placebo group, and the G4 DKD patients showed no signs of heart failure, suggesting that bardoxolone methyl may be a breakthrough agent for improving eGFR in DKD among patients other than those at high risk for heart failure. A Phase 3 Study of Bardoxolone Methyl in Patients with Diabetic Kidney Disease (AYAME) trial is a phase III multicenter, placebo-controlled study in Japan last May 2018 (Fig. 3). Similar to the TSUBAKI study, it will evaluate the efficacy of bardoxolone methyl for DKD. The study is currently underway with a target date of March 2022 and is expected to provide stronger evidence of the renoprotective effect of bardoxolone methyl on DKD.
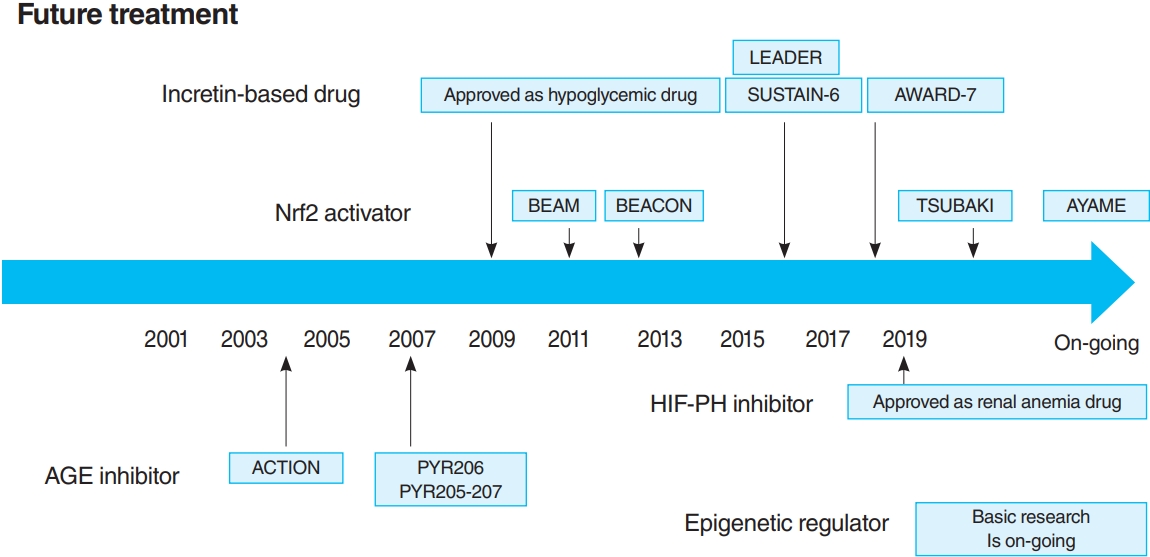
The time course of future treatments. LEADER, the Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results; SUSTAIN-6, the preapproval Trial to Evaluate Cardiovascular and Other Long-term Outcomes with Semaglutide in Subjects with Type 2 Diabetes; AWARD-7, Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease; Nrf2, NF-E2–related factor 2; BEAM, 52-Week Bardoxolone Methyl Treatment: Renal Function in CKD/Type 2 Diabetes; BEACON, the Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes Mellitus: the Occurrence of Renal Events; TSUBAKI, the Phase II Study of Bardoxolone Methyl in Patients with Chronic Kidney Disease and Type 2 Diabetes; AYAME, a Phase 3 Study of Bardoxolone Methyl in Patients with Diabetic Kidney Disease; HIF-PH, hypoxia inducible factor prolyl hydroxylase; AGE, advanced glycation end-product; ACTION, the Aminoguanidine Clinical Trial in Overt Nephropathy; PYR, pyridoxamine.
Presently, the renoprotective effect mechanism of bardoxolone methyl is not fully understood. Nrf2 increases target genes, such as glutathione S-transferase (GST) and NAD(P)H quinone reductase (NQO1), which are detoxifying enzymes, glutathione synthase, heme oxygenase (HO-1), thioredoxin, superoxide dismutase (SOD), and catalase, which are antioxidant enzymes, and inhibits he inflammatory response of macrophages and the activation of nuclear factor-kappa B (NF-κB). These effects may possibly be protective against oxidative stress and chronic inflammation that underlie the progression of DKD. Indeed, diabetic patients have an elevated urinary 8-hydroxy-2’-deoxyguanosine (8-OHdG), an indicator of oxidative stress, which correlates with renal complications [58]. In rats, bardoxolone methyl inhibits angiotensin II-stimulated mesangium contraction [59]. It also stabilizes vascular endothelial cell homeostasis by upregulating nitric oxide synthase (NOS) expression [60]. Furthermore, bardoxolone methyl may ameliorate tubular interstitial damage and inhibit the accumulation of extracellular substrates by suppressing TGF-β [61].
The role of Nrf2 and Keap1 in various models of renal damage has also been investigated by using knockout mice. Nrf2 knockout mice are known to develop lupus nephritis-like autoimmune nephritis after approximately 60 weeks of age, with a deteriorating kidney function [62]. Conversely, Keap1 knockout mice, which degrade Nrf2, were expected to acquire resistance to renal damage by activating Nrf2, but all of them died within 21 weeks of birth due to the hyperkeratinization of the esophagus and other factors [63]. Therefore, for Keap1, studies have been performed in knockdown and conditional knockdown mice, and, in ischemia-reperfusion and nephrotic syndrome models. In NEP25 mice, an improvement in renal damage and an increase in antioxidant factors by Nrf2 activation were noted [64,65]. These experiments in genetically modified mice also show that bardoxolone methyl has a renoprotective effect in activating Nrf2 by suppressing Keap1.
Nrf2 activator has the potential to improve GFR in DKD patients with no high risk for heart failure. However, Nrf2 activator resulted in an increased albuminuria in both BEAM and BEACON studies. Whether this increase is related to glomerular hyperfiltration, which is the pathogenesis of DKD, needs to be examined.
Hypoxia-inducible factor prolyl hydroxylase inhibitor
Regardless of the cause of CKD, irreversible kidney damage is known to progress as CKD progresses beyond a certain point, and the vicious cycle of tubular interstitial hypoxia is thought to be the final common pathway for this progression [66]. In the course of CKD progression, a reduction or loss of blood flow in peritubular capillaries (PTC) is seen from a very early experimental stage. For example, in a rat model of bilateral ischemia-reperfusion injury, a decrease in PTCs has been observed at week 4, and in a mouse model of ischemia-reperfusion injury, fluorescent microangiography has demonstrated a decrease in the perfused area of PTCs at 8 weeks [67]. In addition, the exposure of tubular cells to hypoxia due to reduced blood flow in PTCs leads to the induction of tubular cell apoptosis and cytokines, such as TGF-β, which in turn activate interstitial fibroblasts and increase the production of extracellular matrix, resulting in tubulointerstitial fibrosis progression. Tubulointerstitial fibrosis itself has also been reported to reduce the ability to diffuse oxygen between tubular cells and PTCs [68], creating a vicious cycle, which is thought to be the mechanism of CKD progression. In addition, the vicious cycle of hypoxia also progressed by increased oxygen demand in the nephron caused by hyperfiltration due to a decreased residual glomerular count, decreased NO production and mitochondrial uncoupling due to oxidative stress, and decreased oxygen-carrying capacity due to renal anemia.
Hypoxia-inducible factor (HIF) is a key transcription factor in the response to hypoxia. HIF prolyl hydroxylase (HIF-PH) inhibitors impede the degradation of HIF-α by suppressing HIF-PH, which is responsible for its oxygen-dependent degradation, stabilizing its expression and activating it. HIF activation may have a renoprotective effect by ameliorating this vicious cycle of chronic hypoxia, which has been previously tested in numerous experimental animal models. The long-term observation of ischemia-reperfusion injury (AKI-to-CKD transition model) was reported to reduce renal fibrosis by the administration of HIF-PH inhibitor [69]. In addition, a number of experimental CKD models have demonstrated a reduction in tubular interstitial damage due to HIF activation, including Thy-1 nephritis rats, 5/6 nephrectomy rats, and streptozocin-induced type 1 diabetes mellitus rats. The activation of HIF has been reported to suppress tubular cell apoptosis in Thy-1 nephritis and 5/6 nephrectomy [70,71], whereas its suppression in ischemia-reperfusion models has been reported to increase NF-κB expression and induce inflammation [72], suggesting HIF’s possible renoprotective effect by suppressing cell death and having an anti-inflammatory effect. HIF has also been reported to promote cell regeneration and proliferation; for example, it is known to induce the expression of stromal derived factor-1 and recruit progenitor cells to damaged tissues [73]. Indeed, in five out of six nephrectomized rats, the activation of HIF was effective in preserving PTC [71]. However, with regard to its effects on cell regeneration and proliferation, HIF activation was also reported to induce p27, which represses the cell cycle [74], resulting in tubular cell proliferation of the ischemia-reperfusion injury model [72]. Therefore, whether HIF is protective of tubular cell regeneration remains a debatable point. In this way, HIF has a renoprotective effect in many models of CKD. However, a study on long-term HIF activators in a 5/6 nephrectomy model resulted in worsened renal fibrosis in the long-term (2 to 12 weeks) group [75].
Thus, the long-term effects of HIF-PH inhibition or HIF activation on CKD are likely to be influenced by multiple factors, including primary disease and disease stage, and need to be investigated in ongoing clinical trials and further basic experiments. However, it is notable that a HIF-PH inhibitor significantly reduced albuminuria and glomerular inflammation in a type 2 diabetes mellitus mouse model [76]. As HIF-PH inhibitors have been approved for renal anemia in CKD not yet on dialysis, clinical trials are required to investigate efficacy as CKD treatment, including DKD, in the future (Fig. 3).
Incretin-based drugs (glucagon-like peptide 1 receptor agonists and dipeptidyl peptidase-4 inhibitors)
Incretin, which is secreted from the gastrointestinal tract after food intake, promotes insulin secretion from pancreatic β-cells as blood glucose rises, creating a hypoglycemic effect, and is composed of glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Various extrapancreatic effects of incretin are observed because its receptors are expressed not only in the pancreas but also in the small intestine, kidney, heart, and central nervous system [77]. Dipeptidyl peptidase 4 (DPP4) is capable of degrading GLP-1 and GIP. GLP-1 levels are reported to be low in type 2 diabetes mellitus patients [78], and DPP4 expression is reported to be elevated in kidney biopsy specimens from DKD patients [79].
A recent RCT reported the multiple effects of GLP-1 receptor agonists and DPP4 inhibitors, including renoprotective and hypoglycemic effects. In the Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial [80,81] and the preapproval Trial to Evaluate Cardiovascular and Other Long-term Outcomes with Semaglutide in Subjects with Type 2 Diabetes (SUSTAIN-6) [82], combined renal events (the development of overt albuminuria, serum creatinine doubling and eGFR <45 mL/min/1.73 m2, renal replacement therapy, and kidney disease-related death) were decreased in the GLP-1 receptor agonist treatment group (Fig. 3). However, this reduction was largely due to a reduction in the development of overt albuminuria, with no differences in other parameters (serum creatinine doubling, eGFR <45 mL/min/1.73 m2, and renal replacement therapy). Furthermore, the Exenatide Study of Cardiovascular Event Lowering (EXSCEL) study and the Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial showed a similar reduction in the development of overt albuminuria, but did not inhibit eGFR reduction [83,84]. In dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7) trial, which compared dulaglutide and insulin glargine, even though the glycemic and blood pressure control was comparable in both groups, a decrease in albuminuria as well as a decrease in the rate of eGFR reduction in the dulaglutide group was noted (Fig. 3) [85].
In this way, many RCTs have shown that incretin-based drugs can reduce albuminuria of DKD patients, but there is still controversy on the reduction of the GFR decrease. However, albuminuria is known to be strongly associated with the GFR decrease in DKD, and so, incretin-based drugs are likely to be effective in DKD by reducing albuminuria. In addition, incretin-based drugs are already used for diabetic patients as hypoglycemic agents, and further evidence of their efficacy for DKD is expected to emerge in the future.
AGE inhibitor
AGE is a product of nonenzymatic protein and nucleic acid glycation [86]. It is induced by high blood sugar and oxidative stress and causes damage to various organs. Indeed, AGE accumulation has been reported to correlate with DKD progression in human kidney samples [87,88]. AGE, both directly and through its receptor, receptor of advanced glycation end products (RAGE), causes increased oxidative stress, NF-κB–mediated induction of inflammation, and promotion of fibrosis through the induction of TGF-β [89,90]. The experiments in rats reported that AGE caused DKD symptoms in the kidneys of healthy rats, such as glomerular hypertrophy, mesangial expansion, glomerular basement membrane thickening, glomerular sclerosis, and albuminuria [91]. RAGE overexpression in diabetic mice was reported to worsen DKD histological changes and renal dysfunction progression [92]; conversely, RAGE inhibitor was reported to inhibit DKD progression in rats [93]. In addition, the majority of AGE is eliminated from the kidney [94]. Therefore, the progression of DKD due to AGE forms a vicious cycle.
A number of clinical trials have been conducted on AGE inhibitors, but their effectiveness remains controversial. For example, in pyridoxamine (PYR)-206 and PYR 205/207 studies, pyridoxamine reduced change from baseline in serum creatinine and urinary TGF-β1 excretion after 6 months of treatment in DKD patients (Fig. 3) [95]. The administration of thiamine was reported to reduce the albuminuria of DKD patients [96]. In the Aminoguanidine Clinical Trial in Overt Nephropathy (ACTION) trial of 690 DKD patients, aminoguanidine also reduced proteinuria and inhibited GFR decline (Fig. 3) [97]. On the contrary, the Aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II) trial in DKD patients was discontinued because aminoguanidine was not effective, causing additional side effects such as anemia, liver dysfunction, and vitamin B6 deficiency. When benfotiamine was administered to DKD patients, the albuminuria did not differ from the placebo group [98].
AGE accumulation plays an important role in metabolic memory, and its inhibition is still an attractive treatment. Although some trials have not shown that AGE inhibitors are effective in treating DKD, there is still room for improvement as regards their optimal administration and sample sizes. Large RCT with optimal administration of AGE inhibitors is expected to prove their efficacy in DKD.
Epigenetic regulator
Epigenetics is a DNA sequence-independent regulatory mechanism of gene expression, involving DNA methylation, histone modification, and noncoding RNA. Epigenetic changes, even those caused by transient hyperglycemia or hypoxia, are known to be stored as cellular memory and progressively lead to irreversible renal damage [99]. In fact, vascular endothelial cells exposed to hyperglycemia have been reported to continue to increase oxidative stress and elicit inflammation even after normalization of blood glucose [100]. Furthermore, the aberrant DNA methylation in mesangial cells has been reported to be involved in the progression of diabetic nephropathy [101,102], and in a mouse model of diabetic nephropathy, it, along with altered histone modifications in tubular cells, has been observed [103,104]. Thus, epigenetic genetic changes play an important role in DKD progression.
With regard to DNA methylation, the promoter region of thioredoxin-interacting protein (TXNIP) has been reported to be hypomethylated in whole blood cells and blood monocytes of DKD patients [105]. The knockout of TXNIP was reported to reduce renal damage in the DKD mouse model [106]. Therefore, this hypomethylation may possibly lead to advanced DKD progression. The hypomethylation of runt-related transcription factor 1 (RUNX1) and the increased expression of RUNX1 in tubular cells taken from human kidney biopsy samples [107] were also reported. With regard to histone modification, H3K9 acetylation (H3K9ac) is increased in the promoter region of the high glycated hemoglobin group, according to the report on the histone modification status of blood lymphocytes and monocytes from type 1 diabetes mellitus patients [108]. H3K9ac has been shown to be associated with the Nf-κB pathway, suggesting that increased H3K9ac may be involved in DKD progression by inflammation pathway [6]. Lipids are also known to induce SET7, an H3K4 methyltransferase, and promote the gene expression that causes glomerular hypertrophy and renal fibrosis [109]. The treatment of mesangial cells with losartan under hyperglycemic conditions was reported to reduce the increase of H3K9/14ac in the promoter regions of RAGE, PAI-1, and monocyte chemotactic protein-1 (MCP-1) [110], indicating that AGE and epigenetics are interrelated to produce metabolic memory. HIF-1, a master regulator in the hypoxic response, induces histone-modifying enzymes, such as histone demethylases, including lysine demethylase (KDM) 3A, KDM3B, and KDM3C, to its binding sites, which alter chromatin structure and regulate gene expression [111]. For example, our group showed that a hypoxic stimulus to human umbilical venous endothelial cells caused HIF-1 to bind to the upstream site of glucose transporter 3 (GLUT3) transcriptional initiation point and induced KDM3A, one of the histone demethylases, to this site [112], which meant that hypoxia altered histone modifications and chromatin structures, resulting in the upregulation of gene expressions.
Noncoding RNA is a generic term for RNA other than mRNA that is translated into proteins. It can regulate gene expressions by transcriptional and posttranscriptional mechanisms and has various physiological functions. We found that aspartyl-tRNA synthetase antisense 1 (DARS-AS1), a long noncoding RNA, was induced by HIF1 in tubular cells under hypoxic conditions and that DARS-AS1 inhibited tubular cell apoptosis [113]. As regards DKD, the noncoding RNAs closely related to it include miRNA-21 and miRNA-29. miRNA-21 is highly expressed in the kidney and is known to be closely linked to TGF-β signaling [114,115]. In the blood and urine of transplant patients, peritoneal dialysis patients, and patients with immunoglobulin A (IgA) nephropathy, it was reported to be strongly correlated with renal fibrosis [116-118]. On the contrary, miRNA-29 is considered an antifibrotic factor. Its expression is known to be reduced by TGF-β stimulation in tubular cells [119] and also reduced in unilateral ureteral obstruction (UUO) rats [120]. Thus, numerous miRNAs have been found to act as fibrotic and antifibrotic factors in DKD, but the mechanisms are extremely complex and require further basic research.
Histone modification inhibitors are being studied as a potential treatment for these epigenetic changes. The histone deacetylase inhibitors, such as vorinostat, valproate, sodium butyrate, and trichostatin, have been reported to reduce proteinuria and improve oxidative stress, fibrosis, glomerular damage, and inflammation in the DKD rat model [121-125]. In addition, Dznep, an inhibitor of Ezh2, H3K27 methyltransferase, was reported to inhibit renal fibrosis in mice with UUO and AKI-to-CKD transition [126,127]. Our group showed that AKI damage caused an elevated level of tissue inhibitor of matrix metalloproteinases 2 (TIMP2), which is considered a profibrotic gene, and that TIMP2 was downregulated by Dznep, which meant that TIMP2 was regulated epigenetically and that Dznep recovered this change. Therefore, Dznep can be a novel drug for renal fibrosis via the control of TIMP2. We are now researching on how Dznep regulates TIMP2 epigenetically. Moreover, the administration of MM-102, the inhibitor of myeloid/lymphoid or mixed-lineage leukemia 1 (MLL1), H3K4 methyltransferase, also suppresses fibrosis in AKI-to-CKD transition mice after ischemia-reperfusion injury [128].
CKD, including DKD, is known to be irreversible when it progresses beyond a certain point [60]. If the epigenetic changes in CKD can be elucidated, the potential to develop innovative drugs that can reverse CKD progression is seen. Current research on histone modification inhibitors strongly suggests this possibility. These histone modification inhibitors are expected to show efficacy in clinical trials for the treatment of CKD in the future.
CONCLUSIONS
DKD is the most significant cause of ESKD, which requires renal replacement therapy. However, until recently, RAS inhibitor with multidisciplinary treatments has been the only available treatment option. In 2019, CREDENCE trial proved the efficacy of SGLT2 inhibitors for DKD, adding a new treatment option. However, DKD does not completely inhibit the progression of the disease. Nrf2 activator is a novel drug that improves the kidney function in DKD patients. However, in the BEACON trial, it was discontinued as it caused heart failure; the Japanese TSUBAKI trial showed its effectiveness against DKD. HIF-PH inhibitor has been recently approved for renal anemia and has the potential to be effective against CKD and DKD by enhancing the biological response to hypoxia. Thus, it has the potential to be a breakthrough drug for DKD, like Nrf2 activator. Furthermore, by elucidating new mechanisms of metabolic memory caused by AGE and epigenetic changes in kidney, AGE and histone modification inhibitors are expected to become breakthroughs in DKD treatment.
Notes
CONFLICTS OF INTEREST
Imari Mimura and Tomotaka Yamazaki have no conflict of interest.
FUNDING
Masaomi Nangaku reports personal fees from Akebia, grants and personal fees from Astellas, personal fees from AstraZeneca, grants and personal fees from Bayer, grants and personal fees from Boehringer Ingelheim, personal fees from GSK, grants and personal fees from JT, grants and personal fees from Kyowa Kirin, grants and personal fees from Torii, grants and personal fees from Mitsubishi Tanabe, grants and personal fees from Ono, grants and personal fees from Chugai, grants and personal fees from Daiichi Sankyo, grants from Takeda. Tetsuhiro Tanaka has received honoraria from Kyowa-Kirin and Astellas, and research grant from JT.
Acknowledgements
None