Cell Therapy for Diabetic Neuropathy Using Adult Stem or Progenitor Cells
Article information
Abstract
Diabetic neuropathy (DN) is the most common and disabling complication of diabetes that may lead to foot ulcers and limb amputations. Despite widespread awareness of DN, the only effective treatments are glucose control and pain management. A growing body of evidence suggests that DN is characterized by reduction of vascularity in peripheral nerves and deficiency in neurotrophic and angiogenic factors. Previous studies have tried to introduce neurotrophic or angiogenic factors in the form of protein or gene for therapy, but the effect was not significant. Recent studies have shown that bone marrow (BM)-derived stem or progenitor cells have favorable effects on the repair of cardiovascular diseases. Since these BM-derived stem or progenitor cells contain various angiogenic and neurotrophic factors, these cells have been attempted for treating experimental DN, and turned out to be effective for reversing various manifestations of experimental DN. These evidences suggest that cell therapy, affecting both vascular and neural components, can represent a novel therapeutic option for treatment of clinical DN.
INTRODUCTION
Diabetic neuropathy (DN), a peripheral nervous system disorder, is the most common complication of diabetes mellitus (DM). Currently, about 23.6 million children and adults in the United States (around 8% of the population) are suffering from diabetes. About 60% of long-standing diabetic patients are affected by DM. Patients with DN experience loss of sensation in areas of the body, reduced quality of life as a result of chronic pain, and chronic wounds partly caused by impaired pain sensitivity. Autonomic nerve dysfunction also contributes to decreased quality of life in diabetic patients. DN can affect sensory, motor, and autonomic nerve fibers in any part of the body. The nerves of the lower extremities, having the longest nerve fibers, often develop symptoms first. There are many types of DM syndromes determined by the organ systems and types of nerves affected. Patients may be diagnosed with exclusively sensorimotor or autonomic neuropathy or a combination of both. Severity of symptoms increases gradually over time and correlate with the degree of hyperglycemia [1].
Currently, there are no clinically proven curative treatments for DN. Optimizing blood glucose control and foot care can halt disease progression, but they cannot cure already established nerve damages which usually lead to secondary complications. Symptomatic treatment with pain medications is only partially effective and wounds are difficult to treat. Moreover, deficiency of neurotrophic factors has been regarded as one of the likely mechanisms underlying DN. In a clinical trial, a single treatment of injected neurotrophic cytokines was ineffective for treating DN [2]. However, since DN is characterized by deficiency in angiogenic and neurotrophic factors, cell therapy has recently emerged as an attractive therapeutic strategy for treating DN.
PATHOGENESIS OF DIABETIC NEUROPATHY
Although DN has been widely studied over the past 20 years and its pathology has been established (Fig. 1), the pathogenesis remains unclear. Known pathologies reported in diabetic patients include axonal atrophy, demyelination, nerve fiber loss, and blunted regeneration of nerves. Among many, two well-known pathogenesis was known for DN: metabolic versus vascular. Recent studies, however, have shown that both vascular factors and metabolic interactions are involved at all stages of DN. Erroneous glycemic control is clearly associated with the development of DN: both direct glucose measures and levels of glycated hemoglobin correlate with the occurrence of neuropathy. However, the cause of DN is more complex than dysregulated glucose levels alone. Several contributing factors have been postulated and have received differing degrees of acceptance. Oxidative stress has been considered the final common pathway of cellular injury in hyperglycemia, but the mechanisms leading to DN are more complex, and antioxidants alone do not prevent this disorder. Data from preclinical and clinical studies show that in diabetes, oxidative and nitrosative stress are increased in plasma and tissues [3]. Reduced blood flow through loss of autonomic nerve functions may contribute to the progression of DN, and alterations in microvessels, similar to the pathogenic neovascularization described in diabetic retinopathy and nephropathy.
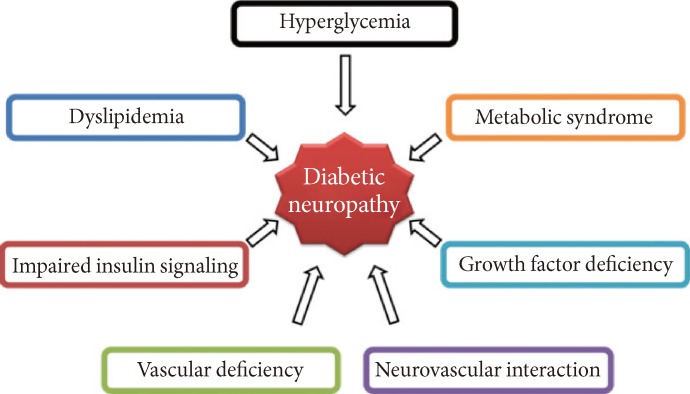
Pathogenesis of diabetic neuropathy (DN). Metabolic interactions vascular factors are involved at all stages of DN. Hyperglycemia, dyslipidemia, metabolic syndrome, impaired insulin signaling, and growth factor deficiency are correlated with the occurrence of neuropathy. Reduced blood flow through loss of autonomic nerve functions may contribute to the progression of DN, and alterations in microvessels, similar to the pathogenic neovascularization described in diabetic retinopathy and nephropathy, also are observed in peripheral nerves.
Hyperglycemia
Excess intracellular glucose is processed by increased flux through one or more glucose metabolism pathways, and prolonged hyperglycemia can lead to cellular damage in several ways. First, excess glycolysis can lead to overload of the mitochondrial electron transport chain and increased generation of reactive oxygen species (ROS). Second, abnormally increased polyol pathway activity increases cellular osmolality, reduces nicotinamide adenine dinucleotide phosphate (NADPH) levels, and leads to oxidative stress. Finally, increased flux through the hexosamine pathway is associated with inflammatory injury [4].
Accumulation of ROS increases lipid, DNA, and protein peroxidation, induces cellular apoptosis, and reduces nerve blood flow (NBF). Increased oxidative stress leads to activation of the the polyADP-ribose polymerase (PARP) pathway, which regulates several inflammatory response gene expressions and causes neuronal dysfunction. On a molecular level, the hyperglycemia is associated with five pathways: the polyol pathway; the advanced glycation end-product (AGE) pathway; the protein kinase C pathway; the PARP pathway; and the hexosamine pathway. Studies suggest that oxidative stress and these five pathways are interdependent and essential to the pathogenesis of neurovascular dysfunction. On a cellular level, hyperglycemia affects sensory, motor, and autonomic neurons by activating the five pathways [5].
In vivo studies show the induction of microvascular ischemia by reducing blood flow results in nerve dysfunction. ROS and reactive nitrogen species are inducing factors of microvascular complications of diabetes. ROS induces impairment of vasodilation of epineural blood vessels, resulting in ischemia to the neural tissue. Oxidative stress also leads to deterioration of Schwann cells, which play a key role as a provider of insulation for neurons, immunologic perineurial blood nerve barrier, and effector of nerve regeneration. Such dysfunction via elevated oxidative stress contributes to the phenotype of DN. Thus, antioxidants have become the therapeutic targets in DN studies. However, only a few studies have suggested that antioxidants can prevent or reverse hyperglycemia-induced nerve dysfunction in experimental DN models [6].
Another consequence of hyperglycemia is the production of AGEs [7], via attachment of reactive carbohydrate groups to proteins, lipids, or nucleic acids. These groups tend to impair the biological function of proteins, thus affecting cellular function. Extracellular AGEs also bind to the receptor for AGE (RAGE), initiating inflammatory signaling cascades, activating NADPH oxidases, and generating oxidative stress. Long-term inflammatory responses are also triggered by the upregulation of RAGE and activation of nuclear factor-kappaB [7].
Dyslipidemia
Dyslipidemia is more likely to occur in patients with type 2 diabetes than in patients with type 1 diabetes. Dyslipidemia is linked to DN, and several underlying mechanisms have been identified. Not only free fatty acids directly cause damage to Schwann cells in vitro, but they also have systemic effects such as promoting inflammatory cytokine release from adipocytes and macrophages. Plasma lipoproteins, especially low density lipoproteins (LDLs), can be modified by oxidation or glycation, and these modified LDLs can bind to extracellular receptors (including the oxidized LDL receptor LOX-1 [lectin-like oxidized LDL receptor-1], toll-like receptor, and RAGE), initiating signaling cascades that activate NADPH oxidase and ultimately lead to oxidative stress. Additionally, cholesterol can be oxidized to oxysterol that may cause apoptosis in neurons [4].
Some evidence exists that neuropathy, particularly when it involves loss of autonomic control of the cardiovascular system, is closely associated with vascular disease factors, including obesity, high plasma levels of cholesterol and triglyceride, and high blood pressure. In a small study of patients with type 1 diabetes, cardiac autonomic neuropathy was associated with impaired ventricular function but not associated with systemic markers of vascular endothelial dysfunction, suggesting that vascular disease itself may not directly lead to neuronal injury. However, a more thorough examination of risk factors and complications in more than 1,400 patients with type 1 diabetes revealed that a decreased vibration perception threshold, which predicts foot ulceration and amputation, was strongly associated with a previous history of cardiovascular disease [8]. Further work is needed to determine whether elevated lipids have direct effects on peripheral neurons and/or Schwann cells.
Studies of patients with type 2 diabetes more frequently demonstrate a correlation between peripheral sensory neuropathy and peripheral vascular disease than studies of patients with type 1 diabetes. In rodents, a high-fat diet leads to accumulation of sorbitol, oxidized lipids and PARP, and activation of lipoxygenases in peripheral nerves before the development of diabetes [9]. Metabolic pathways might mediate neuronal injury in dyslipidemia. Cell culture studies suggest that downstream of inflammation and oxidative and nitrosative stress, protein damage may lead to mitochondrion-mediated activation of cell death mechanisms in neurons. These molecular modifications also activate the endoplasmic reticulum unfolded-protein response in many cell types, which can lead to cell death through endoplasmic reticulum stress [9].
Impaired insulin signaling
Although insulin does not stimulate glucose uptake in neurons, insulin is essential for general neuronal function. Insulin receptors are expressed over neuronal cell bodies in the dorsal root ganglion and peripheral axons sustaining the epidermis [10]. In fact, it was discovered that neuronal insulin receptors are increased after physical injury of peripheral nerves and in diabetes. After locally injecting insulin into the hind paw footpad of diabetic mice, nerve fiber density and mechanical sensation improved [10]. Similarly, intranasal insulin administration in diabetic mice showed a reduction of several pathophysiology and an increase in sensory nerve fibers in the plantar footpads [11].
Insulin has been shown to have neurotrophic effects, promoting neuronal growth and survival. Insulin deficiency (type 1 diabetes) or insulin resistance (type 2 diabetes), which ultimately leads to reduced neurotrophic signaling, is thought to contribute to the pathogenesis of DN. In neurons, insulin resistance occurs by inhibition of the phosphatidylinositide 3-kinase/Akt signaling pathway. Disruption of this pathway can also lead to mitochondrial dysfunction and oxidative stress, further promoting neuropathy [12].
In patients with type 1 diabetes, reduction in C-peptide can lead to nerve dysfunction in several ways, including reduction of sodium potassium adenosine triphosphatase (ATPase) activity, endothelial nitric oxide synthase activity, and endoneurial blood flow. Thus, treatment with C-peptide can slow the progression of neuropathy in patients with type 1 diabetes [13].
The mechanisms outlined above lead to many cellular disturbances, including mitochondrial dysfunction, endoplasmic reticulum stress, DNA damage, and apoptosis. Another layer of complexity is added when considering these processes of cell stress or damage occur in several different cell types within the nerves such as neurons (in axons and at nerve terminals), glial cells, and endothelial cells of the microvasculature. Furthermore, many of these changes trigger the activation and recruitment of macrophages [14], feeding back into inflammatory mechanisms of cell stress and death. Eventually, these different forms of cellular stress lead to dysfunction or death of the nerve, which may develop as clinical neuropathy.
Tight glucose control can reduce neuropathy in patients with type 1 diabetes, but it is not as effective in patients with type 2 diabetes. This disparity is probably related to differences in the underlying mechanisms: hyperglycemia and a reduction in insulin signaling in patients with type 1 diabetes, compared with a combination of hyperglycemia, dyslipidemia, and insulin resistance in patients with type 2 diabetes. The progression of neuropathy between the two types of diabetes may differ due to the difference in duration of proneuropathic changes before the onset or diagnosis of diabetes. Type 2 diabetes does not typically develop rapidly; it occurs after many years of obesity and other aspects of the metabolic syndrome. Tight glucose control will not necessarily reduce dyslipidemia, systemic inflammation, and insulin resistance, and after years of these dysfunctions, neuropathy is difficult to halt or reverse. Although hyperglycemia contributes to the vicious cycles of oxidative stress, inflammation, and cellular damage in patients with type 2 diabetes, reducing hyperglycemia alone might not be enough to stop the cycle [15].
Metabolic syndrome
Rapid advance in diabetes research has increased our knowledge of how components of the metabolic syndrome damage nerves. Along with dyslipidemia and insulin resistance, another principal component of the metabolic syndrome, visceral adiposity, might be particularly detrimental because it causes increased concentrations of free fatty acids in the plasma and also induces a proinflammatory state by secreting adipokines (contributing to the development of insulin resistance). Hypertension, another aspect of the metabolic syndrome, might also be a component contributing to neuropathy, although not much is known about the mechanism. The renin-angiotensin system, which controls blood pressure, is upregulated in obesity, and might contribute to the development of type 2 diabetes (partly through the promotion of insulin resistance and proinflammatory cytokine secretion from adipose tissue) [16]. Angiotensin-converting enzyme (ACE) inhibitors have been shown to improve DN in animal studies, but the underlying mechanism is unclear. Microvascular dysfunction in the nerve and decreased endoneurial perfusion are also thought to contribute to neuropathy [17]. Upregulation of the renin-angiotensin system might also contribute to neuropathy even though this might be regulated by metabolic factors [17].
These mechanisms are probably linked in several levels. Indeed, hyperglycemia, insulin resistance, dyslipidemia, systemic inflammation, and activation of the renin-angiotensin system are all linked together to contribute to a self-perpetuating cycle of oxidative stress, inflammatory signals, and disruption of normal cellular function. At last, these mechanisms interconnect the metabolic syndrome and type 2 diabetes to neuropathy. Thus, neuropathy can possibly develop in the absence of diabetes when other aspects of the metabolic syndrome are activated. One of the main challenges for researchers is to determine which aspects of the network of mechanisms can be blocked to effectively limit or prevent progression of the neuropathy [15].
Growth factor deficiency
In addition to the classical pathogenesis mentioned above, studies have reported the major pathophysiologic role of neurotrophic factors and vascular supply in DN. The two widely considered downstream consequences of the cellular mechanisms are the loss of neurotrophic support and ischemic hypoxia.
Many representative growth factors have dual effects of being both neurotrophic and angiogenic. Some examples are vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and fibroblast growth factor-2 (FGF2, also known as bFGF). Recently, the term angioneurin was coined to refer to a growth factor, which has both angiogenic and direct neurotrophic effects [18]. Studies showed that the levels of these angioneurins are decreased in diabetic animals and are associated with neural function [19,20].
VEGF, a major angiogenic factor, is a potent selective mitogenic cytokine for endothelial cells, and it can be induced by hypoxia through hypoxia-inducible factor (HIF) 1. In ischemic tissues, VEGF induces angiogenesis by stimulating the proliferation and migration of endothelial cells, improving tissue ischemia. VEGF also enhances migration and proliferation of Schwann cells which express kinase insert domain receptor (KDR) or VEGF receptor (VEGFR) 2. It also promotes axonal outgrowth and survival of both the neurons and Schwann cells of superior cervical ganglia and dorsal root ganglia. Like VEGF, IGFs induce vessel remodeling and also have neurotrophic effect. IGFs have been shown to promote neurite outgrowth of neuroblastoma cells and accelerate regeneration of sensory and motor nerves. IGF1 is widely expressed in craniofacial sensory ganglia, sciatic nerve, spinal cord, sensory dorsal root ganglia, and brain, whereas IGF2 is expressed in the brain, vascular structures of the nervous system, and motor neurons. IGF receptors (IGF1R and IGF2R) are present throughout neuronal cell bodies, axons, and nerve terminals. IGF1 expression is reduced in streptozotocin-induced diabetic rats. mRNA and protein expression of both IGF1 and IGF2 are decreased in the nerves of streptozotocin-induced diabetic rats. Similarly, mRNA and protein expression level of IGF1R are decreased in the superior cervical ganglia of streptozotocin-diabetic rats [21]. IGF1 also stimulates Schwann cell mitogenesis and myelination. These effects may be important for interneuronal signaling and peripheral nervous system function. Sonic hedgehog (SHH) modulates embryonic nerve patterning and development. In diabetic animal, SHH mRNA levels are significantly reduced in peripheral nerves. In addition, overexpression of SHH improves blood flow to ischemic nerve and ameliorates nerve function [22]. NGF, a well-known neurotrophic factor, was initially identified as a molecule that promotes survival and differentiation of sensory and sympathetic neurons. However, current studies also show that NGF provides neuroprotective and repair functions. NGF is synthesized by Schwann cells, target cells of sensory and sympathetic neurons such as epithelial cells, smooth muscle cells, and fibroblasts. NGF homozygous knockout mice do not develop proper sympathetic neurons or small neural crest-derived sensory neurons. In addition to these neurotrophic effects, NGF directly induces angiogenesis [23].
Vascular deficiency
Maintaining adequate blood supply to nerves is crucial in maintaining nerve structure and function. Deficiencies in the blood supply to neural tissues through vasa nervorum and blood vessel within peripheral nerves largely contribute to pathogenetic mechanism of DN. Several mechanisms regarding vascular structural changes in ischemia on diabetic nerve have been proposed. The most common abnormality in the structure of diabetic vasa nervora is the thickening of basement membrane, which is highly correlated with neuropathic severity [24]. In addition, decrease in nerve conduction velocity (NCV) in diabetic rats is preceded by impaired vasodilation in epineurial arterioles, which is partly mediated by ROS production [25]. In contrast to constricted epineurial arteriole, endoneurial capillaries appear to have a variable patency. Luminal areas of endoneurial capillaries were increased in rodent and feline models of DN. However, those in human showed mixed results.
Studies of blood vessel number or density in the nerves of diabetic subjects also showed mixed results. In human, the endoneurial capillary density was reported to be higher in early diabetic patients than healthy subjects. Conversely, the endoneurial capillary density of diabetic patients with established neuropathy showed no significant difference to that of healthy subjects. However, recent studies have shown decreased functional capillary density using lectin perfusion, which is a method for measuring capillary density [19]. This discrepancy in the number of endoneurial capillaries may arise from different methods or markers used in examining capillaries. Studies altogether suggest that the number of capillary increases in response to ischemia in early diabetic condition, and the number, particularly the functional capillaries, decreases due to impaired neovascularization under prolonged diabetic condition [19].
Despite some controversies on the structural aspects, it is clear that DN is accompanied by ischemia and hypoxia of microcirculatory nutrient vessels in nerves [26]. A vicious pathogenic cycle may develop due to the regulated microcirculation by humoral, endothelial, and neural factors. Microcirculatory dysfunction results in peripheral nerve dysfunction which results in abnormal regulation of the microcirculation, ultimately leading to overall nerve dysfunction. Treatment with various vasodilatory agents, such as prostaglandin E1 analogues, alpha-adrenergic receptor blockers, ACE inhibitors, angiotensin II receptor antagonists, and endothelin receptor antagonists in animal models of diabetes has shown to reduce the endoneurial blood flow [26].
Further evidence for the impaired vascularization and ischemia in DN shows that there is a decrease in factors that promote or maintain blood vessel formation in DN (such as VEGFs, angiopoietins, IGFs, and NGF). This observation led to studies of local delivery of the angiogenic factors VEGF-A and VEGF-C into diabetic rats that resulted in increased vasa nervora density and NCVs. Additionally, VEGFs have direct neurotrophic effects that may contribute to the amelioraton of NCVs. For instance, angiogenic medications such as statins have shown to improve nerve functions in DN [26].
Neurovascular interaction
One of the main pathogenic factors in the development of DN is reduced NBF. Various in vitro and in vivo experiments evidently show that amelioration of NBF improves nerve functions. Studies reported a decrease in endoneurial blood flow on diabetic patients and in presence of hypoxia. Direct measures of nerve perfusion evidently show that decreased sural NBF is strongly associated with DN [27].
Neurotrophic factors promote the innervation of target tissues during development. For instance, target organs release NGF to secure the survival of sympathetic and sensory neurons. Neurotrophic factors, such as BDNF, neurotrophin 3 (NT3) and NT4, ciliary neurotrophic factor (CNTF), and members of the glial cell line-derived neurotrophic factor (GDNF) family, also promote the survival of various neuronal populations in the developing nervous system [28].
Consequently, deficiency of these neurotrophic factors can cause neurodevelopmental disorders [28]. Diabetes reduces BDNF, NGF, and NT3 in peripheral nerves by limiting anterograde and retrograde axonal transport. Intrathecal delivery of NGF or NT3 improves myelinated fiber innervation in the dermal footpad of diabetic mice, and thus, lack of neurotrophic support can affect fiber morphology. Initial discovery of neurotrophic factors has shown that some of these molecules may regulate angiogenesis. Genetic studies in mice show that BDNF is essential factor in maintaining cardiac vessel-wall stability during development. Binding to TrkB receptor of endothelial cells, BDNF stimulates angiogenesis in the heart, skeletal muscle, and skin. The binding recruits proangiogenic bone marrow (BM) cells to stimulate revascularization of ischemic limbs. NT4 also has similar activity as it binds to TrkB receptor [29]. NGF also stimulates angiogenesis in two ways. It stimulates indirectly by increasing the expression of VEGF and directly by promoting vascular cell growth. NT3 (through binding to TrkC) and leukemia-inhibitory factor both serve as inhibitors of the growth of some endothelial cells, whereas GDNF and CNTF have no effect on angiogenesis.
Many common angiogenic factors have recently been discovered to have neuronal activity. Among many factors, evidence indicates that VEGF, one of the main regulators of vessel growth in all tissues, also affects many neuronal and glial cell types by binding to VEGFRs. Study revealed that hypoxia upregulates the expression of VEGF through the binding of HIFs to a hypoxia-response element in the promoter of its gene [30]. Other VEGF family members, such as VEGFB and VEGFC, also have direct effects on neurons, but the roles for these proteins in neurodegeneration have not been discovered. Platelet-derived growth factors (PDGFs) are the closest homologues of the VEGF family members, and these two families even share a common ancestral origin in fruit flies and worms. PDGF receptors (PDGFR) are activated to stabilize nascent vessels by recruiting smooth-muscle cells around endothelial channels. Interestingly, PDGF receptor is not required for adult-neuron survival in healthy conditions, but it is neuroprotective in pathological conditions [31].
Recent genetic studies reveal effects of other angiogenic factors in neurodegeneration. Angiogenic properties of angiogenin (ANG) are mediated by direct effects on endothelial cells. In addition, ANG directly stimulates the survival of motor neurons [32]. There are also a number of other angiogenic factors, such as angiopoietin 1 (ANG1), that regulate neurogenesis or neuroprotection, but currently, there is no genetic evidence supporting effects of angioneurins in neurodegeneration. Other molecules, such as FGF2, epidermal growth factor (EGF), transforming growth factor 1, hepatocyte growth factor, IGF1, erythropoietin, and others, have also been implicated in postnatal neuron loss or adult-onset neurodegeneration. Dysfunctional mutations of these factors result in a spectrum of histopathological features, including neuron loss, loss of function, and degeneration, that develop either spontaneously or after injury [32].
Quiescent endothelial cells require survival signals to cope with stressful conditions. Genetic and pharmacological studies show that a low level of VEGF is required to maintain the integrity of quiescent vessels [33]. This supports the hypothesis that endothelium-specific loss of VEGF causes generalized vessel disintegration with resultant brain infarcts and bleeding [33], and it could also explain why VEGF protects vessels in peripheral nerves against damage by chemotherapeutic agents. FGFs and probably many more angioneurins have similar 'vasculo-protective' effects on quiescent vessels.
Demyelination is a characteristic of DN. Several angioneurins promote (re)myelination through direct effects on oligodendrocytes, Schwann cells, and their precursors. In vitro, various angioneurins exert mitogenic effects on oligodendrocytes and Schwann cells, and also protect those cells from apoptosis. Studies show Pdgfa knockout mice that survived birth develop tremor due to severe hypomyelination of neuronal projections, whereas heterozygous platelet-derived growth factor receptors (PDGFR) deficient mice develop impaired oligodendrocyte-progenitor proliferation and oligodendrocyte regeneration in adult models of toxin-induced demyelination [34]. The type 1 IGF receptor has a critical role in remyelination; it stimulates the proliferation of oligodendrocyte progenitors. In a model of ischemic peripheral neuropathy, VEGF preserves axon myelination and promotes axon regeneration through the direct stimulation of Schwann-cell migration and proliferation [35].
CELL THERAPIES FOR DIABETIC NEUROPATHY
Growth factors are attractive therapeutic option for DN because they can promote neuron survival and functional integrity, as well as repair of damaged nerves. Some growth factors are angiogenic, and their therapeutic effects are mediated by blood vessel growth that supply nutrients and oxygen to nerves. Other growth factors, such as NT3, are neurotrophic, and their therapeutic effects are via promoting neural regeneration and survival. Growth factors, known as angioneurins (VEGF, FGF2, NGF, BDNF, IGF1), have both angiogenic and neurotrophic properties. The power of these growth factors in the treatment of DN was shown by Schratzberger et al. [26]. They were the first to inject VEGF encoding plasmids into rat and rabbit models of diabetes. The VEGF-treated animals showed normalization in NCV, increase in angiogenesis of vasa nervora, and increase in nerve fiber density. When the plasmid VEGF was applied to human patients, mild, but statistically significant symptomatic improvement was observed in a randomized, double-blinded trial [36]. However, the authors also reported that VEGF therapy was associated with adverse side effects that did not reach statistical significance. As this study has a relatively small sample size, further study is required to conclusively determine the effects of plasmid VEGF therapy. Other growth factors, such as IGF1 and IGF2 have also been studied in animal models of DN and have shown protective effects against development of neuropathy independent of changes in blood glucose. FGF2 promotes angiogenesis and neurogenesis.
As mentioned, emerging evidence has indicated that angiogenic factors such as VEGF-A, VEGF-C, SHH, and statin restore microcirculation in the affected nerves accompanied by functional improvement [37]. On the other hand, lack of neurotrophic factors has been regarded as an important pathogenic mechanism of DN. Administration of neurotrophic factors such as NGF, IGF1 and IGF2, CNTF, or GDNF was shown to ameliorate DN in animal models [5].
These findings suggest that a therapeutic modality which can target both angiogenic and neurotrophic processes may have more value in treatment of DN. In this sense, cell therapy using stem or progenitor cells has advantages over single gene or protein therapy. Cell therapy can increase multiple angiogenic and neurotrophic factors and potentially supplement specific type of cells required for vascular or neuronal regeneration. Currently, various circulating or BM cells were shown to have favorable effects for treating DN. An advantage of using circulating or BM-derived cells is that they can be harvested from a patient's own peripheral blood (PB) or BM, and reintroduced back to the patient (Fig. 2).
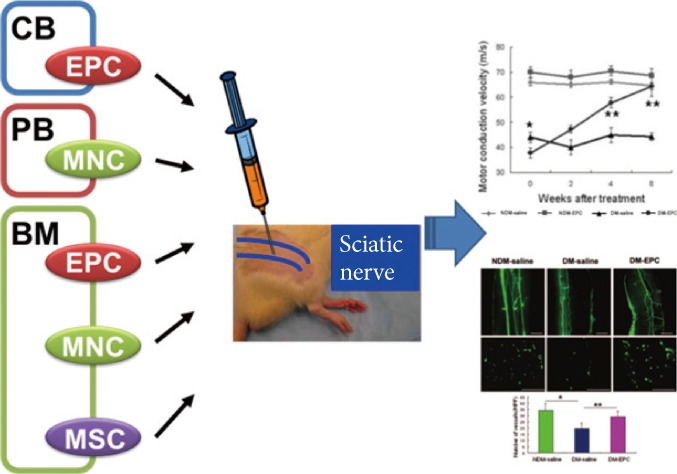
Cell therapy for diabetic neuropathy (DN) using adult stem or progenitor cells. Candidate adult stem or progenitor cells include mononuclear cells (MNCs), endothelial progenitor cells (EPCs), or mesenchymal stem cells (MSCs) from cord blood (CB)-, bone marrow (BM)-, or peripheral blood (PB)-derived cells. Through angiogenic and neurotrophic effects, these cells can reverse various functional and histologic manifestations of DN.
Cord blood derived endothelial progenitor cells
EPCs can be isolated from the BM, cord blood, and PB. Many experimental studies revealed that EPCs have a potent ability for neovascularization and that the transplantation of EPCs improves tissue ischemia EPCs isolated from cord blood have a greater proliferative potential and a higher cell cycle rate than EPCs from other sources, suggesting that cord blood-derived EPCs may more effectively contribute to therapeutic vasculogenesis [38].
Naruse et al. [39] have demonstrated that therapeutic neovascularization using human umbilical cord blood-derived EPCs reversed DN. EPCs were isolated and expanded on day 7 of culture from cord blood mononuclear cells (MNCs). Highly proliferative cord blood-derived EPCs were maintained even in the diabetic condition, claiming that EPCs from cord blood have a better proliferative potential and a higher cell cycle rate than EPCs from other sources [39]. Unilateral intramuscular injection of human cord blood-derived EPCs into hindlimb skeletal muscles significantly ameliorated impaired sciatic motor NCV and sciatic NBF in the EPC-injected side of streptozotocin-induced diabetic nude rats compared with the saline-injected side of diabetic nude rats. Histological study revealed an increased number of microvessels in hindlimb skeletal muscles in the EPC-injected side of diabetic rats. These findings suggest that transplantation of EPCs from cord blood may be a useful treatment for DN. If one can avoid adverse immunological reaction to allografts, transplantation of cord blood-derived EPCs with high proliferative capacity may have an effective treatment of human DN [39].
Peripheral blood mononuclear cells
Blood flow was shown to be improved by implantation of hematopoietic MNCs in ischemic myocardium and hindlimb muscles [40]. Therapeutic angiogenesis resulting from implantation of blood cells also ameliorated diabetic peripheral neuropathy [41]. PB-derived MNCs (PB-MNCs) contain considerably less EPCs than BM-derived MNCs (BM-MNCs), and implantation of PB-MNCs was less effective in ischemic regions than BM-MNCs [42]. However, implantation of PB-MNCs was still associated with a significant improvement in collateral perfusion and regional function probably as a consequence of supplying angiogenic factors to the ischemic regions [40].
The recovery of NBF and motor NCV (MNCV) in the streptozotocin-induced diabetic rats was almost equivalent to implantation of either PB-MNCs or BM-MNCs [41], despite the BM-MNC fraction containing approximately 10 times more EPCs than the PB-MNC fraction. Moreover, immunohistochemical study revealed that no significant increase in vessel numbers in the sciatic nerve resulted from implantation of either cell fraction. This finding suggests that these implantations improve NBF and MNCV as a result of an arteriogenic effect of angiogenic factors on muscles rather than angiogenesis in the sciatic nerve [41].
PB-MNC and BM-MNC fractions contained similar quantities of VEGF and FGF2, whereas IL1B and TNF levels were higher in PB-MNCs than in BM-MNCs [40]. These studies indicated VEGF may play a key role in angiogenesis in ischemic tissues. VEGF-neutralizing antibody abolished the effects of both PB-MNCs and BM-MNCs implantations [41], suggesting that VEGF may also be responsible for the observed increase in sciatic NBF. It has been reported that PB-MNCs synthesize and release VEGF as a proinflammatory reaction [43], and consequently, inflammatory PB-MNCs themselves may have a more important role in VEGF production than EPCs, resulting in equivalent effects following implantation of either PB-MNC or BM-MNC fractions. Notwithstanding this limitation, as PB-MNCs and BM-MNCs fractions are obtained routinely from donors and then implanted into patients with hematopoietic diseases, autologous implantation of PB-MNCs and BM-MNCs fractions may also provide a safe therapeutic strategy for the treatment of diabetic peripheral neuropathy.
Bone marrow mononuclear cells
BM-MNCs are derived from BM and isolated using density gradient centrifugation. BM-MNCs are heterogeneous cell population including lymphocytes, hematopoietic stem/progenitor cells, EPCs, and mesenchymal stem cells (MSCs). BM-MNCs have been shown to augment neovascularization by increasing a broad range of angiogenic factors, including FGF2, VEGF, and angiopoietin 1 in the tissue [44,45]. In animal models, transplantation of BM-MNCs into ischemic limbs [45] and myocardium [44] increased neovascularization and collateral blood vessel formation. These effects of BM-MNCs have also been documented in patients with limb ischemia in randomized controlled trials [46]. BM-MNCs are easily isolated and do not have to be expanded by ex vivo culture. This ease of isolation makes BM-MNCs an attractive source of cells for therapeutic neovascularization.
Recent studies have shown favorable therapeutic effects of BM-MNCs on experimental DN. Hasegawa and colleagues [41] showed that implantation of either PB-MNCs or BM-MNCs in a rat model of DN improved motor NCV and blood flow around the sciatic nerve, which is possibly mediated by VEGF secreted from MNCs. This study suggests that BM-MNCs are more effective than PB-MNCs as BM-MNCs include significantly more EPCs than PB-MNCs. Recently, Kim et al. [20] reported that intramuscularly transplanted BM-MNCs preferentially localize to the nerves in diabetic rats, especially around vasa nervorum, and increase expression of various angiogenic and neurotrophic factors in the nerves. The vascularity of these nerves improved and NCV levels were almost normalized [20]. These studies suggest that the vasa nervorum may play a pathogenetic role in both the development and reversal of DN. This study further suggested that angiogenesis is the central mechanism of BM-MNC-induced neovascularization in experimental DN because, from their observation, BM-MNCs do not differentiate into, nor fuse with, endothelial cells in the nerves at a detectable level.
More recently, other reported study also showed the transplantation of freshly isolated BM-MNCs alleviated neuropathic pain in the early stage of streptozotocin-induced diabetic rats [47]. Mechanical hyperalgesia and cold allodynia in SD rats were measured as the number of foot withdrawals to von Frey hair stimulation and acetone application, respectively. The BM-MNC transplantation significantly ameliorated mechanical hyperalgesia and cold allodynia in the BM-MNC-injected side. Furthermore, the slowed MNCV/SNCV and decreased sciatic NBF (SNBF) in diabetic rats were improved in the BM-MNC-injected side. BM-MNC transplantation improved the decreased mRNA expression of NT3 and number of microvessels in the hind limb muscles. There was no distinct effect of BM-MNC transplantation on the intraepidermal nerve fiber density [47]. The immunohistological study revealed that the BM-MNC transplantation increased the number of microvessels in the ipsilateral soleus muscle. In the sciatic nerve, STZ-induced diabetes induced a reduction of the NBF and this deficit was recovered by BM-MNC transplantation. Kim and the colleagues [20] indicated that transplanted BM-MNCs preferentially engrafted in the sciatic nerve and improved NBF. These results, therefore, suggest that improvement of the blood flow in the tissues including nerve vessels is one of the crucial effects of BM-MNC transplantation. This study showed that transplantation of BM-MNCs into the unilateral hindlimb skeletal muscles inhibited mechanical hyperalgesia in the ipsilateral side but not in the contralateral side [47]. Two clinical studies supported these observations linking painful DN with alterations in blood flow, and demonstrated significant benefits of pain relief from the use of vasodilators, isosorbide dinitrate spray, and glyceryl trinitrate patches [48]. These results with the previous studies [20,39] supported showing that autologous transplantation of BM-MNCs could be useful for the treatment of painful DN.
Bone marrow-derived mesenchymal stem cells
MSCs have been reported to secrete various cytokines that exhibit angiogenic and neurosupportive effects. MSCs reside in the BM stromal fraction, which provides the cellular microenvironment supporting hematopoiesis. Because MSCs have an adherent nature, they are expandable in culture, and it is relatively easy to obtain a sufficient number of cells for cell therapy. MSCs have recently been shown to differentiate into multilineage cell types and to secrete various cytokines, including FGF2 and VEGF. Through these actions, transplantation of MSCs has been experimentally reported to be a promising strategy for the treatment of ischemic diseases such as myocardial infarction and arteriosclerosis obliterans. Shibata and the colleagues [49] showed that MSC transplantation ameliorated DN. MSCs were isolated from BM of adult rats and transplanted into hind limb skeletal muscles of STZ-induced diabetic rats by unilateral intramuscular injection. Four weeks after transplantation, VEGF and FGF2 productions in transplanted sites, current perception threshold, NCV, SNBF, capillary number to muscle fiber ratio in soleus muscles, and sural nerve morphometry were significantly increased in MSC-injected thigh muscles of STZ-induced diabetic rats. Furthermore, colocalization of MSCs with VEGF and FGF2 in the transplanted sites was confirmed. STZ-induced diabetic rats showed hypoalgesia, delayed NCV, decreased SNBF, and decreased capillary number to muscle fiber ratio in soleus muscles, which were all ameliorated by MSC transplantation. Sural nerve morphometry showed decreased axonal circularity in STZ-induced diabetic rats, which was normalized by MSC transplantation. These results suggest that MSC transplantation could have therapeutic effects on DN through paracrine actions of growth factors secreted by MSCs [49].
Bone marrow-derived endothelial progenitor cells
The development of vascular system consists of two processes: vasculogenesis and angiogenesis. Vasculogenesis refers to the de novo formation of blood vessels from EPCs or angioblasts that differentiate into endothelial cells, whereas angiogenesis is growth of pre-existing vasculature by sprouting of new capillaries through proliferation and migration of endothelial cells. Until recently, vasculogenesis was thought to be restricted to embryonic development, while angiogenesis was considered to be responsible for neovascularization in embryos and adults. This view was challenged with the discovery of BM-derived EPCs, which circulate in adult PB, home to ischemic tissue and incorporate into foci of neovascularization, leading to de novo blood vessel formation.
The identity of EPCs is complicated by the complexity of the definition and various methods to define the cells. It is now apparent that different subsets of peripheral or BM derived cells, including hematopoietic stem cells, monocytes and circulating endothelial cells, can differentiate into endothelial-like cells. BM-derived EPCs in the adult PB express a subset of hematopoietic stem cell markers. Specifically, CD34, CD133, and VEGF receptor-2 have been proposed as candidate markers for human EPCs [50]. However, there are no known specific markers to identify EPCs without cultivation. Ex vivo expanded human EPCs express various endothelial cells markers such as CD31, CD34, KDR, vascular endothelial (VE)-cadherin, bind lectins, and incorporate Dil-acetylated LDL. The origin of EPCs is further obscured by the two distinctive types of EPCs arising from different culture methods. "Early EPCs," are mainly derived from MNCs or monocytes and do not proliferate after a few weeks. On the other hand, "late EPCs" form colonies after more than 2 weeks in culture, have cobblestone morphology, and rapidly proliferate [51]. The distinctive characteristics of these two types of EPCs are reinforced by the different expression of cell surface markers. Early EPCs express pan-leukocyte and monocytic/macrophage markers such as CD45, CD11b, and CD14 while late EPCs do not. Early EPCs are also therapeutically effective in vivo while evidence for therapeutic efficacy of late EPCs are limited to date [51].
Endothelial differentiation of EPCs and whether this differentiation plays a main role in the therapeutic benefit of EPCs in recovering damaged tissue function is controversial. Several recent studies have demonstrated differentiation of EPCs into endothelial lineage cells with incorporation into blood vessels [52]. However, other investigators claim that BM-derived cells including EPCs do not undergo endothelial differentiation nor incorporate into vessel walls [53]. These discrepancies may be due to the difference in cell types, the use of different animal models or the rigor of the criteria to define endothelial differentiation.
As mentioned above, cord-blood derived EPCs were effective for treating DN [39]. This study claimed that mechanistically, the therapeutic effects are due to the increased differentiation of EPCs into endothelial cells in hindlimb muscles, which then led to an increase in SNBF. However, this study did not demonstrate the fate of the EPCs in tissues, nor did it address the mechanisms by which transplanted EPCs increase neovascularization in muscles or nerve. Given that most recent studies have argued against the endothelial differentiation of EPCs as a major mechanism for neovascularization, endothelial differentiation does not appear to underlie such magnitude of therapeutic effects toward DN [54].
However, a study by the author's group reported that local transplantation of BM-derived EPCs improved various manifestations of experimental DN through dual angiogenic and neurotrophic effects on peripheral nerves [19]. This study uncovered several important mechanistic role of EPCs on DN [19]. First, intramuscularly injected EPCs exert therapeutic effects through direct modulation of nerves, not through muscular neovascularization. Second, the therapeutic effects of EPCs are mainly mediated by humoral factors, rather than the direct endothelial differentiation. Third, the vasa nervora density in the sciatic nerves was augmented following the EPC treatment. Fourth, intramuscularly injected EPCs preferentially recruited to sciatic nerves, preferentially localized in close proximity to vasa nervora, and infrequently differentiated into endothelial cells [19]. A majority of engrafted EPCs survived in peripheral nerves for at least 12 weeks and sustained expression of angiogenic and neurotrophic factors. Fifth, EPC transplantation increased proliferation and decreased apoptosis of endothelial and Schwann cells.
The most unique finding was the direct effect of EPCs on peripheral nerves. The study was the first to demonstrate that EPCs induce neovascularization directly in nerves [19]. Angiogenesis could have played a more important role than vasculogenesis in the increase of neural neovascularization. This neural angiogenesis occurred through upregulation of various angiogenic factors in nerves after EPC transplantation. Various paracrine factors including VEGF-A, FGF2, BDNF, SHH, and stromal cell derived factor-1α were expressed in the peripheral nerves. These factors have synergistic effects on angiogenesis and neuroprotective effects. In fact, this study was the first to demonstrate unambiguous dual angiogenic and neurotrophic effects of EPCs. This upregulation of various biological factors may be the critical benefits that cell therapy can have over any single protein or gene therapy, enabling the synergistic effects of multiple neuroangiogenic factors necessary for neurovascular recovery.
Histologically, the author's study also uncovered novel engraftment and retention characteristics of BM-derived cells in tissues [19]. Following a series of reports on the short-term engraftment of any BM cells in a myocardial infarction model [55], the prevailing notion was that adult stem/progenitor cells could not sustain their engraftment more than a few weeks. However, the study by Jeong et al. [19] clearly rebutted this notion that BM-EPCs could survive more than 12 weeks in nerves. While the EPCs which were directly injected into the hindlimb muscles disappeared mostly in the muscles within 8 weeks, the EPCs robustly survived for more than 12 weeks in the sciatic nerves. Interestingly, the study by Naruse et al. [39] showed that capillary density, which had decreased in hindlimb muscles of diabetic rats at 12 weeks of diabetes, was significantly increased after cord-blood EPC treatment. However, this study indicated that blood flow and capillary density in hindlimb were not significantly changed after EPC treatment. This long-lasting cell retention is compatible with the observation that EPCs homed to peripheral nerves far more preferentially than to muscles. This magnitude of interaction between any BM cells and steady-state tissues was not previously demonstrated in other tissues or organs. This series of studies with EPCs or BM-MNCs strongly argue that the engraftment characteristics of BM cells depends more on the recipient environment than on the transplanted cells themselves [19,20]. This evidence supports that despite the controversy of EPCs on blood vessel forming capability in certain models like myocardial infarction [54], it is evident that EPCs can play an important role in vessel homeostasis and revascularization [56]. The distinct properties of BM-derived EPCs such as peripheral neurotropism, long-term retention, and vascular localization of EPCs worked together to exert prolonged paracrine or humoral effects and reversal of various functional and pathologic manifestations of DN [19,20,39,41].
CONCLUSIONS
Diabetes injures peripheral nerves in various distributions. The most common pattern is distal sensory polyneuropathy (DSP), which is characterized by numbness, tingling, pain, or weakness that affects the nerves in a stocking and glove pattern, beginning in the distal extremities. DSP leads to substantial pain, morbidity, and impaired quality of life. Societal, personal, and healthcare costs associated with DN are high. Unfortunately, few interventions are available for the remediation of nonpainful symptoms, and glucose control is the only proven disease-modifying intervention for these patients. Although pain is a common feature, it is often under-reported and undertreated. However, many effective therapies exist for DNP, including medicines designed to treat seizures and depression.
Many areas of research into DN are yet to be fully explored, but there are promising lines of investigation that could lead to improved prevention and treatment of the disorder. The magnitude of the effect of glucose control on neuropathy is much smaller in patients with type 2 diabetes than in those with type 1 diabetes. In view of this small effect size and the fact that many patients with type 2 diabetes continue to develop neuropathy despite adequate glucose control, discovery of modifiable risk factors for neuropathy is essential. Components of the metabolic syndrome, including prediabetes, are potential risk factors for neuropathy, and studies are needed to establish whether they are causally related to neuropathy. These lines of enquiry will have direct implications for the development of new treatments for DN.
DN is a systemic and progressive disease and its manifestations can take many years to develop. Cell therapy may not be a standard treatment option for all stages of DN because different stages of DN are marked by different structural or functional changes. At present, cell therapy may be applied to those patients who suffer from intractable symptoms, acute exacerbation, or combined diseases such as diabetic foot ulcers or critical limb ischemia.
Practically, as the safety of autologous BM-derived cells has been documented by a number of clinical trials [57], it is highly recommended to advance this strategy into a pilot clinical trial for those who are severely affected by DN. Particularly, EPCs will be effective in treating DN when combined with diabetic wounds or peripheral vascular obstruction as the therapeutic effects were already shown in these conditions. However, there are a few remaining concerns in cell therapy strategy. First, the effectiveness of the patient's own cells needs to be evaluated considering the possibility that BM cells derived from diabetic subjects may be impaired in therapeutic potential. Experiments using the autologous cells derived from diabetic subjects are necessary to address these concerns. However, although the efficacy of autologous diabetic cells is less potent, there may be still ways to overcome these defects to a certain extent. One strategy is to enhance their angiogenic and neurotrophic effects by culturing cells and activating necessary pathways with small molecules or growth factors. Second, the long-term effects of cell therapy need to be evaluated. Given that DN is a disease progressing over a long time, a single injection of cells may not be enough to maintain the nerve function over a long period of time. One approach is to implant cells repeatedly to maintain their effects. At present, the duration of the beneficial effects of cell therapy in DN is unknown and a critical issue that requires further investigation. In many cases, the first manifestation of DN is a diabetic foot or ulcer which sometimes requires an amputation and a long-term care and significantly reduces patients' quality of life. Cell therapy in this case can be critical to rescue further tissue loss.
Cardiovascular autonomic neuropathy (CAN) is associated with increased risk of cardiovascular morbidity and mortality in diabetic patients [58]. Although CAN is one of the most frequently studied complications of diabetes [59] and cell therapy has been reported to be effective in improving ischemic cardiovascular disease [56] and peripheral neuropathy, cell therapy has yet been evaluated in either animal models or human patients with CAN. Future studies are required to determine the effects of cell therapy in CAN.
Future directions of cell therapy for DN will take steps toward enhancing the potency of candidate cells, using both gene and cell therapy, and working with combination of various cell types such as those derived from induced pluripotent stem (iPS) cells. Once generated, iPS cells can offer a plentiful and renewal source of cells that can be induced to differentiate into cells of interest [60]. Conclusively, cell therapy may become an innovative alternative therapeutic option for treating advanced DN. However, further research is necessary to overcome some limitations and possible adverse effects of cell therapy.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants 1DP3DK094346; HHSN268201000043C (Program of Excellence in Nanotechnology Award); NSF-EBICS (Emergent Behaviors of Integrated Cellular Systems) grant, CBET-0939511 and ACTSI pilot grant (PHS grant UL1 RR025008 from the CTSA program, NIH, and NCRR).
Notes
No potential conflict of interest relevant to this article was reported.