Chemokine Systems Link Obesity to Insulin Resistance
Article information
Abstract
Obesity is a state of chronic low-grade systemic inflammation. This chronic inflammation is deeply involved in insulin resistance, which is the underlying condition of type 2 diabetes and metabolic syndrome. A significant advance in our understanding of obesity-associated inflammation and insulin resistance has been recognition of the critical role of adipose tissue macrophages (ATMs). Chemokines are small proteins that direct the trafficking of immune cells to sites of inflammation. In addition, chemokines activate the production and secretion of inflammatory cytokines through specific G protein-coupled receptors. ATM accumulation through C-C motif chemokine receptor 2 and its ligand monocyte chemoattractant protein-1 is considered pivotal in the development of insulin resistance. However, chemokine systems appear to exhibit a high degree of functional redundancy. Currently, more than 50 chemokines and 18 chemokine receptors exhibiting various physiological and pathological properties have been discovered. Therefore, additional, unidentified chemokine/chemokine receptor pathways that may play significant roles in ATM recruitment and insulin sensitivity remain to be fully identified. This review focuses on some of the latest findings on chemokine systems linking obesity to inflammation and subsequent development of insulin resistance.
INTRODUCTION
Excess adiposity increases the risk of developing a variety of pathological conditions, including type 2 diabetes, cardiovascular disease, steatohepatitis, and several types of cancer [1-4]. In the past two decades, advances in obesity research have led to the recognition that adipose tissue is an endocrine organ that secretes hormones or cytokines termed adipokines [5,6]. Adipokines play crucial roles in the regulation of appetite and satiety control, energy expenditure, insulin sensitivity and insulin secretion, endothelial function, and blood pressure [3,6]. The adipokines leptin and adiponectin, which are primarily secreted by adipocytes, play a major role in the pathogenesis of obesity and its comorbidities, including type 2 diabetes and cardiovascular disease [7,8]. On the other hand, obesity involves a state of chronic low-grade systemic inflammation. Recently, areas of active investigation focus on the molecular bases of metabolic inflammation and potential pathogenic roles in diabetes and cardiovascular disease [2,9,10]. This inflammation develops in response to an excess of nutrient flux into metabolic tissues including adipose tissue, liver, and skeletal muscle and is currently recognized as an important link between obesity and insulin resistance. Obesity-associated systemic inflammation is characterized by increased circulating concentrations of proinflammatory cytokines and chemokines, and activation of several kinases that regulate inflammation, including c-Jun NH2 terminal kinase, IκB-kinase β/nuclear factor κB and mammalian target of rapamycin/S6 kinase that interfere with insulin action in adipocytes and hepatocytes.
Increasing evidence supports that obesity-induced inflammation is mediated primarily by immune cells such as the macrophages and T lymphocytes in metabolic tissues. In particular, a significant advance in our understanding of obesity-associated inflammation and insulin resistance has been recognition of the critical role of adipose tissue macrophages (ATMs). ATMs are a prominent source of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, that can block insulin action in adipocytes via autocrine/paracrine signaling and cause systemic insulin resistance via endocrine signaling, providing a potential link between inflammation and insulin resistance [10-12]. In both humans and rodents, ATMs accumulate in adipose tissue with increasing body weight and their content correlates positively with insulin resistance [13-15]. Importantly, tissue macrophages are phenotypically heterogeneous and have been characterized according to their activation/polarization state as M1 (or "classically activated" proinflammatory macrophages) or M2 (or "alternatively activated" noninflammatory macrophages [16-18]). M2 ATMs predominate in lean mice, whereas obesity induces the accumulation of M1 ATMs with high expression of TNF-α, IL-6 and inducible nitric oxide synthase [17], leading to a proinflammatory environment in white adipose tissue (WAT). Thus, both recruitment and proinflammatory activation of ATMs is required for the development of insulin resistance in obese mice.
Chemokines, a family of cytokines that induce leukocyte chemotaxis, are deeply involved in the development of allergic diseases and autoimmune diseases. More than 50 chemokines which exhibit various physiological and pathological properties have been discovered to date [16,19]. Recent studies have shown that preadipocytes and adipocytes express chemokines, and have demonstrated the infiltration of bone marrow-derived macrophages into obese adipose tissue, which is involved in the development of insulin resistance. This review summarizes various roles of chemokine and chemokine receptor (chemokine system) in inflammation, and also focuses on some of the latest findings on the chemokine systems that link adipose tissue inflammation with the pathology of insulin resistance.
CLASSIFICATION OF CHEMOKINES AND CHEMOKINE RECEPTORS
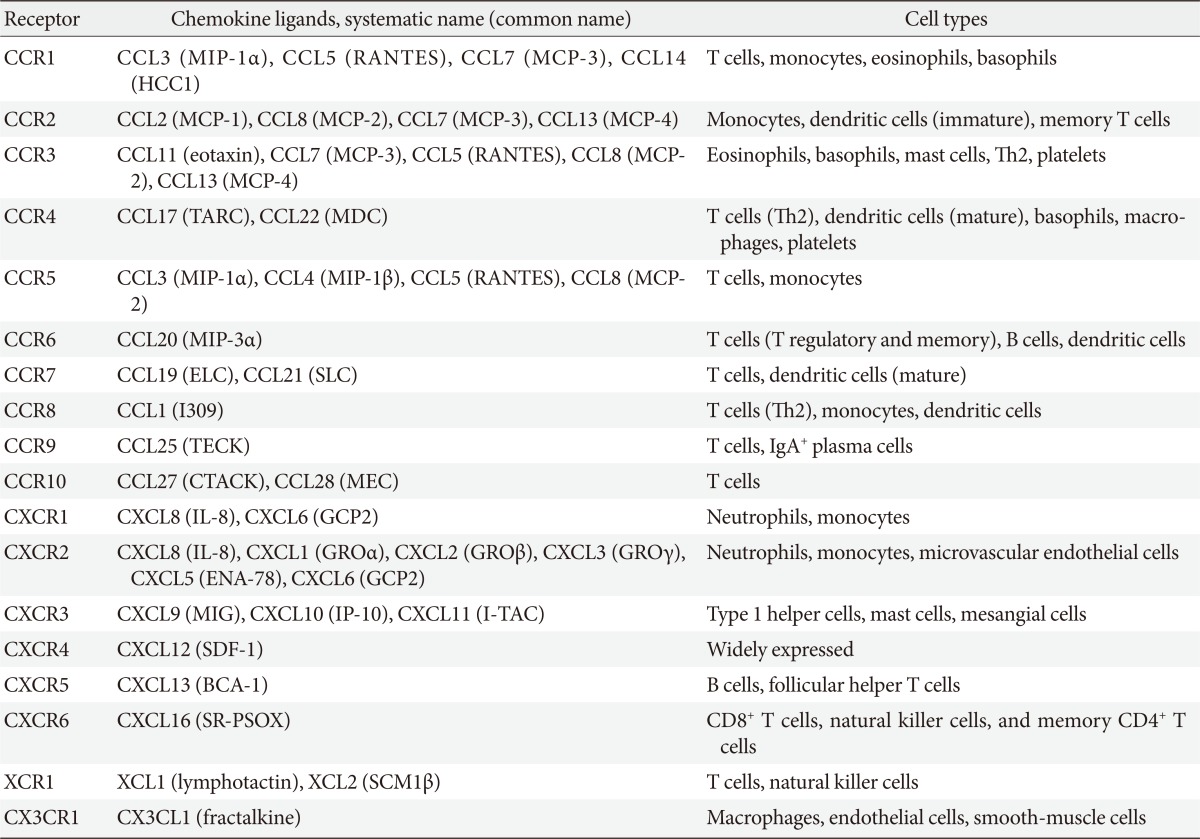
Chemokines attract leukocytes to areas of inflammation and lesions and play a key role in leukocyte activation. Chemokines were first discovered as cytokines that are chemotactic for neutrophils and monocytes. Ever since, many studies have been conducted to identify the roles of chemokines in acute, neutrophil-predominant inflammation and chronic, monocyte- and lymphocyte-predominant inflammation [20,21]. To date, more than 50 chemokines have been identified in humans (Table 1). Chemokines have four conserved cysteine residues. Based on the motif patterns involving two N-terminal cysteine residues, chemokines can be classified into the following four subfamilies: CXC, CC, C, and CX3C (where X is any amino acid residue) (Table 1) [16,19]. The CXC chemokines are mainly chemotactic for neutrophils and are known for their involvement in acute inflammation whereas most of CC chemokines act on monocytes, T cells, eosinophils, and basophils, which mediate chronic inflammation and allergy [16,19].
Chemokines and chemokine receptors
All chemokines signal via seven-transmembrane G-protein-coupled receptors (or chemokine receptors). To date, 18 chemokine receptors have been identified, including 11 CC chemokine receptors, 6 CXC chemokine receptors, and one for each C (XCR1) and CX3C chemokine receptor (Table 1). Most chemokines bind to several chemokine receptors and chemokine receptors have overlapping ligand specificities [16,19]. Although some chemokines have a one-to-one specificity (specific receptors), most chemokines bind to the same receptor (shared receptors) (Table 1) [16,19]. For example, four chemokine ligands, including monocyte chemoattractant protein (MCP)-1, MCP-2, MCP-3, and MCP-4 bind to C-C motif chemokine receptor 2 (CCR2) receptor. Even when multiple ligands interact with a single receptor, diverse effects are produced by different ligands because the binding affinity and the resulting effect differ across ligands. Furthermore, as chemokines are differently expressed, distributed, and regulated in cells and tissue, they may play different roles in physiological conditions or diseases.
INFLAMMATORY CHEMOKINES AND HOMEOSTATIC CHEMOKINES
Although chemokines appear to exhibit a high degree of functional redundancy, the functions of the individual chemokines can be classified into two types: 'inducible' or 'inflammatory' and 'constitutive' or 'homeostatic' [16,19]. Inflammatory CXC and CC chemokines promptly recruit neutrophils, monocytes and eosinophils to the site of injury or inflammation, cluster on chromosomes 4 and 17, and share the same receptor [22]. Furthermore, a number of inflammatory chemokines act together as a potent inducer of cell migration (quantitative regulation). Since the late 1990s, it has been clearly demonstrated that several chemokines exhibit different properties from those of inflammatory chemokines. These homeostatic chemokines act specifically and constantly on lymphocytes and dendritic cells, and are characterized by relatively specific ligand-receptor pairs [19,22]. It is believed that homeostatic chemokines modulate the physiological homing of naive lymphocytes and dendritic cells to secondary lymphoid tissue, and that they are involved in highly specific migration control (qualitative control).
CHEMOKINE SYSTEMS AND DISEASE
There have been great advances in chemokine research through the elucidation of the potential pathogenic roles of inflammatory chemokines in allergic diseases and autoimmune diseases, including bronchial asthma, rheumatoid arthritis, and inflammatory bowel disease. In such diseases, chemokines and their receptors promote the onset and modify the severity of each of these diseases through the selective accumulation of leukocytes in the site of inflammation. MCP-1, a prototype of the chemokine [23,24], as well as MIP-1α and MIP-1β are the CC chemokine subfamily that are isolated from monocyte and macrophages, and have been shown to be closely linked with the development of many inflammatory diseases. The signals of CC chemokine receptors CCR1, CCR2, and CCR5 are also involved in the pathogenesis of multiple sclerosis, characterized by chronic inflammation induced by monocytes, macrophages, and T cells (Table 1) [19,22]. In acute respiratory distress syndrome, on the other hand, CXC chemokines act on neutrophils, causing a large number of neutrophils to infiltrate into the lung, thus triggering a severe acute inflammatory reaction. Furthermore, in recent years, increasing evidence has shown that chemokines and their receptors play pathogenic roles on cancer, cardiovascular disease, neurodegenerative disorder, and metabolic diseases which have led many researchers to explore the role of chemokines on various other diseases associated with chronic inflammation.
PIVOTAL ROLE OF MCP-1-CCR2 IN OBESITY-INDUCED INSULIN RESISTANCE
It has become increasingly evident that chemokine system also involves chronic subacute inflammation that is the common underlying condition of obesity, insulin resistance, and type 2 diabetes. The interaction of MCP-1 with its receptor CCR2 is considered pivotal in obesity-induced insulin resistance. Several groups have reported that mice with targeted deletions in the genes for Mcp-1/Ccl2 and its receptor Ccr2 have decreased ATM content, decreased inflammation in fat, and protection from high-fat (HF) diet-induced insulin resistance [25,26]. Conversely, mice overexpressing MCP-1 in adipose tissues have increased numbers of ATMs along with insulin resistance [25,27]. Therefore, the MCP-1-CCR2 axis is of central importance for promoting ATM recruitment and insulin resistance in mice. More recent studies, however, have produced conflicting results and indicated greater complexity than suggested by earlier reports. Loss of MCP-1 neither attenuates obesity-associated macrophage recruitment to WAT nor improves metabolic function, suggesting that MCP-1 is not critical for obesity-induced ATM recruitment and systemic insulin resistance [28,29]. Furthermore, although Ccr2-/- mice fed a HF diet have fewer macrophages in WAT compared with WT mice [26] CCR2 deficiency does not normalize ATM content and insulin resistance to the levels in lean animals, indicating that ATM recruitment and subsequent insulin resistance are also regulated by MCP-1-CCR2 independent signals.
This complexity and redundancy of chemokine signaling may account for these conflicting results. In fact, other chemokine systems have also been implicated in ATM infiltration in obese mice [30-32]. Indeed, serum levels of CXCL5 produced by ATM are increased in obese mice and humans, and mice lacking CXCR2, the receptor of CXCL5, show resistance to the onset of obesity-induced disorders of glucose metabolism [32]. Moreover, previous research has suggested that the concentration of keratinocyte, which is homologous to human IL-8, increases in the adipose tissue and plasma in an obese model, and that bone marrow chimera mice with bone marrow cells from CXCR2 deficient mice show decreased obesity-induced inflammation and insulin resistance compared to controls [31]. However, additional unidentified chemokine/chemokine rereceptor pathways that may play significant roles in ATM recruitment and insulin sensitivity remain to be fully identified.
CCR5 LINKS OBESITY TO INSULIN RESISTANCE BY REGULATING BOTH MACROPHAGE RECRUITMENT AND M1/M2 STATUS
We recently identified and characterized a critical role for CCR5, a different CC chemokine receptor, in the regulation of the adipose tissue inflammatory response to obesity and the development of insulin resistance (Fig. 1) [33]. We made several important observations. First, expression of CCR5 and its ligands is significantly increased and is equal to that of CCR2 and its ligands in the WAT of obese mice, particularly in the macrophage fraction. Second, fluorescence-activated cell sorter analysis clearly demonstrates a robust increase in CCR5+ ATMs in response to a HF diet even after normalizing for stromal vascular cell number and fat weight. Third, and most important, Ccr5-/- mice are protected from insulin resistance, hepatic steatosis, and diabetes induced by HF feeding. It is noteworthy that two distinct models, both Ccr5-/- mice and chimeric mice lacking CCR5 only in myeloid cells, are protected from HF diet-induced hyperinsulinemia and glucose intolerance through a reduction in ATM accumulation. Finally, it is interesting that an M2-dominant shift in ATM is induced in obese Ccr5-/- mice. Therefore, we conclude that deficiency of CCR5 causes an M2-dominant phenotypic shift in ATMs, which contributes to the attenuation of obesity-induced insulin resistance.
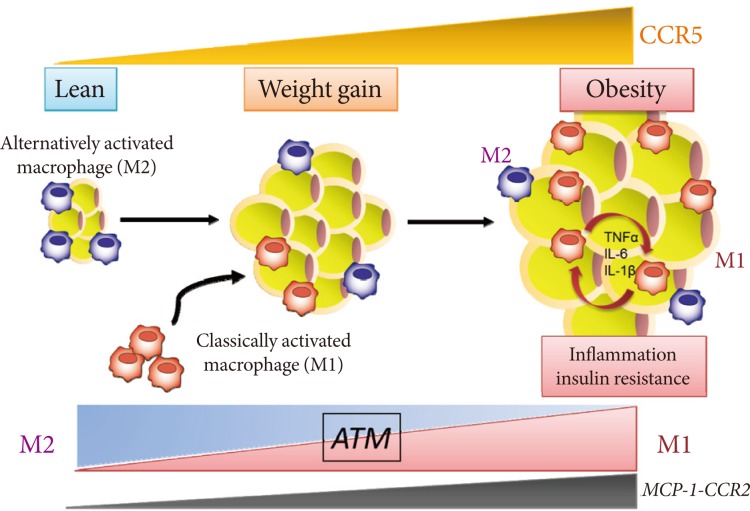
C-C motif chemokine receptor 5 (CCR5) promotes obesity-induced inflammation and insulin resistance. Kitade et al. [33] recently identified and characterized a role for CCR5, a CC chemokine receptor, and made several important observations. First, expression of CCR5 and its ligands is significantly increased and is equal to that of CCR2 and its ligands in white adipose tissue (WAT) of obese mice. Second, CCR5+ adipose tissue macrophages (ATMs) accumulate in WAT of obese mice. Third, Ccr5-/- mice are protected from insulin resistance and diabetes through both reduction in ATM accumulation and induction of an alternatively activated, M2 dominant shift in ATM. Taken together, these data indicate that CCR5 provides a novel link between obesity, adipose tissue inflammation, and insulin resistance. TNF, tumor necrosis factor; IL, interleukin; MCP, monocyte chemoattractant protein.
Our study provides new information about the role of CCR5, a new chemokine system, in obesity-induced insulin resistance in an animal model [33]. It is important that the effects of CCR5 do not appear to result from global alterations in adipocyte biology. Thus, decreased ATM recruitment does not appear to be secondary to changes in adiposity because the adipocyte size of obese Ccr5-/- mice and age-matched controls is similar. Moreover, expression of adipocyte-derived factors such as leptin and adiponectin in WAT and plasma levels are similar between genotypes. Additionally, a bone marrow transplantation study revealed that lack of CCR5 expression in macrophages alone was sufficient to protect mice from the HF diet-induced insulin resistance; this was associated with a marked reduction in ATM infiltration. These data support the conclusion that CCR5+ ATMs are important in the development and maintenance of obesity-induced adipose tissue inflammation and insulin resistance in mice. Recent human studies have also shown upregulation of the expression of not only MCP-1-CCR2 but also other CC chemokines (CCL5, CCL7, CCL8, CCL11) and their receptors (CCR1, CCR3, CCR5) in the visceral fat of morbidly obese individuals in whom macrophage infiltration has been confirmed [34]. Taken together, CCR5-mediated signals in the adipose tissue may be involved, in some way, in the induction and maintenance of obesity-induced inflammation and in the development of insulin resistance in both rodents and humans.
CCR5 AS A NOVEL PLAYER IN THE ADIPOSE TISSUE INFLAMMATION AND INSULIN RESISTANCE
Do the two CC chemokine receptors, CCR2 and CCR5, play common or unique roles in obesity-induced adipose tissue inflammation and insulin resistance? Importantly, no significant compensatory increase in the expression for CCR2, or vice versa, has been found. Therefore, CCR5, independently from and/or cooperatively with CCR2, plays a role in the maintenance of ATM dysfunction and insulin resistance once obesity and its metabolic consequences have been established (Fig. 1). Moreover, similar to the case in Ccr5-/- mice, HF diet-induced increased fat mass are minimally affected by Ccr2 deficiency, and obese Ccr2-/- mice matched for adiposity with controls showed reduced ATM recruitment and improved systemic insulin sensitivity [26]. Therefore, the effects of either CCR5 or CCR2 do not appear to result from global alterations in adipocyte biology. However, HF feeding promotes accumulation of M1 macrophages in WAT of WT mice, whereas increase in M1 ATMs are markedly suppressed in Ccr5-/- mice [33]. In contrast, M2 expression within ATMs is increased in Ccr5-/- mice on a HF diet, suggesting that deficiency of CCR5 causes an M2-dominant phenotypic shift in ATMs, which contributes to the attenuation of obesity-induced insulin resistance. Interestingly, such a phenotypic switch is not observed in Ccr2-/- mice [17].
CCR5 is preferentially expressed on Th1 cells [35]. Recent studies have demonstrated that obesity is associated with increased accumulation of not only macrophages but also T cells in adipose tissue. Wu et al. [36], showed that RANTES/CCL5 mRNA levels are highly correlated with the T cell marker CD3 in human visceral adipose tissue. However, the numbers of CD3+ T cells, CD4+ T cells and CD8+ T cells did not differ in WAT of HF diet-fed WT and Ccr5-/- mice [33], suggesting that CCR5 deficiency affects ATM recruitment more prominently. One important question concerns whether the loss of CCR5 affects the M1/M2 status in the bone marrow or peripheral blood (Fig. 2). In mice, two major distinct subsets of blood monocytes have been reported: Ly6Chi and Ly6C- monocytes. The former, called proinflammatory/classical monocytes, preferentially accumulate in atherosclerotic plaques and exhibit a strong inflammatory response to lipopolysaccharide [37]. In contrast, the latter, known as resident/remodeling/patrolling monocytes, participate in the resolution of inflammation [38]. Both Ly6Chi and Ly6C- monocytes are recruited to sites of inflammation or injury (Fig. 2) [39]. Although the relationship between the monocyte subtypes and their fate as M1/M2 macrophages remains unknown, that loss of CCR5 could cause alteration of Ly6Chi and Ly6C- monocytes subsets at the level of either bone marrow or peripheral blood, and that this contributes to the M2-dominant shift of ATM in obese Ccr5-/- mice.
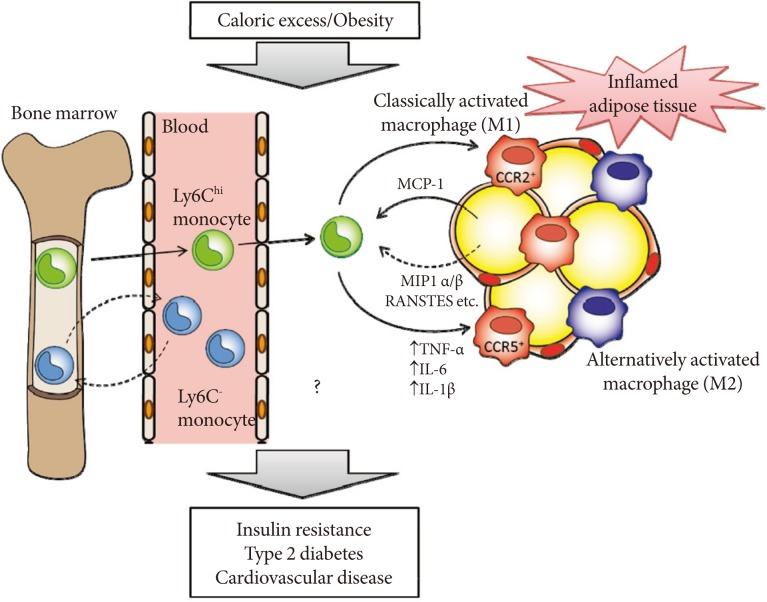
Hypothetical role of C-C motif chemokine receptor 5 (CCR5) and monocyte chemoattractant protein (MCP)-1-CCR2 in the development of adipose tissue inflammation and insulin resistance in obesity. Obesity causes both the recruitment and proiflammatory activation of adipose tissue macrophages (ATMs). Adipocytes or preadipocytes begin to secrete MCP-1 as well as other chemokines, such as MIP1α, MIP1β, and RANSTES (ligands for CCR5) in obesity. Thereafter, CCR2+ and/or CCR5+ macrophages accumulate and presumably maintain the inflammation as M1 or classically activated macrophages in obese adipose tissue. Ly6Chi monocytes exit the bone marrow in a CCR2-dependent manner and are recruited to inflamed tissues. CCR5 may also regulate recruitment of Ly6Chi and Ly6C- monocytes and their fate as M1/M2 ATMs. Once these ATMs are present and active, cytokines (tumor necrosis factor [TNF]-α, interleukin [IL]-6, and IL-1β) are produced. Therefore, CCR5, independently from and/or cooperatively with CCR2, could play an important role in the induction and maintenance of obesity-induced inflammation and insulin resistance.
Our study provides new information regarding the role of CCR5 as a novel link among obesity, adipose tissue inflammation, and insulin resistance (Fig. 1). However, many questions have yet to be answered, including how CCR5 and its ligands are induced in response to either a HF diet or obesity; how CCR5 regulates M2 macrophages; which metabolic tissue/organ is responsible for enhanced insulin sensitivity in Ccr5-/-; and of the 50 chemokines in metabolic diseases, what distinct roles are played by CCR5? As cytokines and chemokines work in networks, the effect of individual molecules on metabolic function depends on their place in the hierarchy of the network. Several recent reports suggest that obesity increases macrophage infiltration to insulin target organs other than adipose tissue. For example, MCP-1-mediated infiltration of CCR2+ bone marrow cells was observed in the liver of the ob/ob mice, and adenovirus-induced overexpression of MCP-1 in the liver of wild-type mice treated with HF diet was found to increase lipogenesis and to promote fatty liver disease [40]. Chemokine-mediated macrophage infiltration into the liver might be therefore related with the pathology of not only insulin resistance but also steatohepatitis. In light of these new data, however, CCR5 may be a promising therapeutic target for insulin resistance, metabolic syndrome, and type 2 diabetes. Further work is required to gain a systematic understanding of how CCR5 and MCP-1-CCR2 as well as other chemokine systems, connect obesity, inflammation, and insulin resistance.
CONCLUSIONS
Vigorous research has been conducted to explore the physiological functions of chemokines in controlling inflammatory and immune response, as well as to elucidate their pathophysiological roles in the development of autoimmune diseases and allergic diseases. It has also become increasingly evident that chemokines play an important role in obesity-induced insulin resistance and its comorbidities, including type 2 diabetes and cardiovascular disease. MCP-1-CCR2 as well as other chemokine systems may be deeply involved not only in tissue- and organ-level inflammation caused by interaction between adipose tissue and macrophages, but also in the onset of localized inflammation caused by cross-talk between adipose tissue and other organs and the subsequent development of systemic insulin resistance. There are many questions, however, that have yet to be answered, including how chemokine expression is regulated in obesity, how chemokines regulate macrophage polarization, and what the different roles of over 50 chemokines are in metabolic diseases. Further research on chemokines in relation to the molecular bases of metabolic inflammation and pathogenic roles in insulin resistance is expected to contribute to the development of new drugs that could control inflammation- or immune-mediated disorders such as metabolic syndrome and type 2 diabetes.
ACKNOWLEDGMENTS
This work was supported by the following grants to Tsuguhito Ota: Program for Improvement of Research Environment for Young Researchers from the Special Coordination Funds for Promoting Science and Technology; Grant-in-Aid for Young Scientists (B) (22790854) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Uehara Memorial Foundation; Kanae Foundation for the Promotion of Medical Science; Japan Heart Foundation/Novartis Grant for Research Award on Molecular and Cellular Cardiology; Banyu Life Science Foundation International; Research Grant of Astellas Foundation for Research on Metabolic Disorders; Japan Research Foundation for Clinical Pharmacology and Daiichi-Sankyo Foundation of Life Science. The author thanks Dr. Mayumi Nagashimada for drawing the figure.
Notes
No potential conflict of interest relevant to this article was reported.