- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 34(4); 2010 > Article
-
ReviewO-GlcNAc Modification: Friend or Foe in Diabetic Cardiovascular Disease
- Udayakumar Karunakaran1, Nam Ho Jeoung2
-
Korean Diabetes Journal 2010;34(4):211-219.
DOI: https://doi.org/10.4093/kdj.2010.34.4.211
Published online: August 31, 2010
- 3,302 Views
- 35 Download
- 16 Crossref
1Department of Medical Sciences, Kyungpook National University School of Medicine, Daegu, Korea.
2Department of Fundamental Medical and Pharmaceutical Sciences, Catholic University of Daegu CU Leaders' College, Gyeongsan, Korea.
- Corresponding author: Nam Ho Jeoung. Department of Fundamental Medical and Pharmaceutical Sciences, Catholic University of Daegu CU Leaders' College, 5 Geumnakro, Hayang-eup, Gyeongsan 712-702, Korea. syjeoung@cu.ac.kr
Copyright © 2010 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- CHARACTERIZATION OF O-GLCNACYLATION OF PROTEINS
- O-GLCNACYLATION, DIABETES, AND INSULIN RESISTANCE
- O-GLCNACYLATION INDUCES VASCULAR DYSFUNCTION
- O-GLCNACYLATION ON VASCULAR SMOOTH MUSCLE CELLS
- O-GLCNACYLATION ON ENDOTHELIAL DYSFUNCTION
- O-GLCNACYLATION AND CARDIOVASCULAR PROTECTION
- CONCLUSIONS
- REFERENCES
ABSTRACT
- O-Linked β-N-acetyl glucosaminylation (O-GlcNAcylation) is a dynamic post-translational modification that occurs on serine and threonine residues of cytosolic and nuclear proteins in all cell types, including those involved in the cardiovascular system. O-GlcNAcylation is thought to act in a manner analogous to protein phosphorylation. O-GlcNAcylation rapidly cycles on/off proteins in a time scale similar to that for phosphorylation/dephosphorylation of proteins. Several studies indicate that O-GlcNAc might induce nuclear localization of some transcription factors and may affect their DNA binding activities. However, at the cellular level, it has been shown that O-GlcNAc levels increase in response to stress and augmentation of this response suppresses cell survival. Increased levels of O-GlcNAc have been implicated as a pathogenic contributor to glucose toxicity and insulin resistance, which are major hallmarks of type 2 diabetes and diabetes-related cardiovascular complications. Thus, O-GlcNAc and its metabolic functions are not yet well-understood; focusing on the role of O-GlcNAc in the cardiovascular system is a viable target for biomedical investigation. In this review, we summarize our current understanding of the role of O-GlcNAc on the regulation of cell function and survival in the cardiovascular system.
- The addition of O-linked β-N-acetyl glucosamine (O-GlcNAc) to the hydroxyl groups of serine and threonine residues on target proteins is a dynamic regulatory post-translational modification of nuclear and cytosolic proteins [1]. It is involved in a wide range of biological processes such as nuclear transportation, transcription and translation, signal transduction, cytoskeletal reorganization, proteasomal degradation, and apoptosis [2,3]. In terms of abundance and protein distribution, O-GlcNAc is often considered to be analogous to the phosphorylation of proteins [3]. Just as protein phosphorylation is regulated by hundreds of kinases and phosphatases, O-GlcNAcylation is also tightly regulated by an enzymatic process. So far, only O-GlcNAc transferase (OGT) has been shown to catalyze the O-GlcNAcylation of proteins and one enzyme, N-acetyl-glucosaminidase (O-GlcNAcase or OGA), catalyzes its removal [4,5]. The end-product of the hexosamine biosynthesis pathway (HBP), uridine diphosphate-N-acetyl glucosamine (UDP-GlcNAc), acts as a substrate for the O-GlcNAcylation of proteins [6]. Approximately 2-5% of the total intracellular glucose enters the HBP and is ultimately converted to UDP-GlcNAc. The metabolism of glucose via the HBP is essential for the synthesis of O-GlcNAc, which serves as a metabolic sensor that attenuates the cellular response to extracellular stimuli based on the energy state of the cell [7]. Several studies have demonstrated that an acute increase in O-GlcNAc synthesis improves cell survival, while a sustained increase in O-GlcNAc levels has been implicated as a pathogenic contributor to glucose toxicity and insulin resistance, major hallmarks of type 2 diabetes (T2D) and diabetes-related cardiovascular complications [8].
- In this review, we summarize our understanding of the role of O-GlcNAc in mediating both the adverse effects of diabetes as well as cellular protective mechanisms in the cardiovascular system.
INTRODUCTION
- Under normal conditions, two to five percent of the glucose taken up by cells is consumed by the HBP, and entrance of glucose into the HBP is regulated by glutamine: fructose-6-phosphate amidotransferase (GFAT), which converts fructose-6-phosphate to glucosamine-6-phosphate using glutamine as an amine donor. Glucosamine-6-phosphate is then metabolized via various intermediates, leading to the synthesis of UDP-GlcNAc [9]. In addition, UDP-GlcNAc can be also increased by addition of exogenous glucosamine, which enters into cells via the glucose transporter system and is phosphorylated to glucosamine-6-phosphate by hexokinase, instead of the GFAT-dependent HBP, leading to an increase in UDP-GlcNAc levels [10]. Using UDP-GlcNAc as a substrate, OGT catalyzes the transfer of GlcNAc via an O-linkage to specific serine and threonine residues on target proteins [11]. OGT activity/O-glycosylation is vital for life, confirmed by the observation that deletion of the OGT gene was embryonic lethal [12]. The level of O-GlcNAcylation of nucleocytosolic proteins is also regulated by O-GlcNAcase, which catalyzes the removal of the sugar moiety from the target proteins. Similar to the processes of phosphorylation/dephosphorylation, O-GlcNAcylation on the target proteins rapidly activates/deactivates the activities of proteins involved in many aspects of cellular processing. At the same time, O-GlcNAcylated proteins can also be modified via phosphorylation, which modulates the functions of proteins by influencing protein-protein interactions and protein localization (Fig. 1) [13].
CHARACTERIZATION OF O-GLCNACYLATION OF PROTEINS
- Insulin resistance plays a major pathophysiological role in T2D and is tightly associated with major health problems, including obesity, heart diseases, and dyslipidemia [14]. Marshall et al. [15] demonstrated that flux through the HBP is responsible for the development of insulin resistance and remarked that the development of insulin resistance requires glucose, insulin, and glutamine. The role of glutamine in the development of insulin resistance has been found to occur via the regulation of GFAT, the rate limiting enzyme in the HBP [16]. Inhibition of GFAT with either azaserine or 6-diazo-5-oxo-norleucine abrogated the effects of hyperglycemia on the development of insulin resistance [17]. Studies have shown that an increase in cellular UDP-GlcNAc and O-GlcNAcylation levels on target proteins due to high glucose and glucosamine treatments leads to oxidative stress and endoplasmic reticulum stress, which have been shown to cause chronic inflammation and insulin resistance [18]. Blocking of the removal of O-GlcNAc using the OGA inhibitor, (2-acetamido-2-deoxy-D-glucopyranosylidene) amino-N-phenyl carbamate (PUGNAC) results in a decrease in insulin-response glucose uptake in 3T3-L1 adipocytes [19]. In insulin target cells, there appears to be a negative feedback regulation of glucose transport via the flux of glucose through the HBP [20]. Insulin resistance and the corresponding decrease in glucose uptake are correlated with a defect in the translocation of the glucose transporter GLUT4 to the plasma membrane [20]. Recent studies point to phosphatidylinositol-4, 5-bisphosphate (PIP2)-assisted remodeling of filamentous actin at the inner leaflet of the plasma membrane (cortical F-actin) as another crucial step in insulin-stimulated GLUT4 translocation [21]. Bhonagiri et al. [22] showed that exposing 3T3-L1 adipocytes to excess HBP flux leads to a reduction in PIP2 content in the plasma membrane, with a concomitant loss in cortical F-actin, which affects insulin-induced GLUT4 translocation into the plasma membrane. In addition to the effects on GLUT4 translocation, it has been reported that there are associations among HBP, insulin resistance, and impairment of Akt/PKB signaling [23]. Yang et al. [24] demonstrated that upon insulin stimulation, OGT is rapidly recruited from the nucleus to the plasma membrane by PIP3 through a region adjacent to the catalytic domain II at the carboxyl terminus of the OGT and demonstrated a potential mechanism by which Akt is presumably able to be O-GlcNAcylated by the PIP3-recruited OGT. Based on these findings, impairment of O-GlcNAcylation regulation is reflected by a decrease in glucose uptake through insulin resistance.
O-GLCNACYLATION, DIABETES, AND INSULIN RESISTANCE
- Hyperglycemia and insulin resistance are major causative factors for T2D and its vascular complications. High circulating glucose concentrations alter functions of the vascular endothelial cells and the underlying smooth muscle cells [25]. Hyperglycemia can induce vascular complications via several different mechanisms, and one of the mechanisms is an increase of the HBP flux of glucose and O-GlcNAcylation of target protein [8]. Hyperglycemia-induced oxidative stress inhibits glyceraldehydes 3-phosphate dehydrogenase, resulting in a decrease in glycolytic flux and an increased glucose flux in the HBP [26]. This increased flux of glucose may influence vascular remodeling as it can modulate the cell proliferation or cell death of different vascular cell types, such as pericytes, endothelial cells, or smooth muscle cells.
O-GLCNACYLATION INDUCES VASCULAR DYSFUNCTION
- Vascular smooth muscle cell (VSMC) dysfunction is a major risk factor of diabetic cardiovascular disease. VSMCs are highly specialized cells whose principal functions are the contraction of blood vessels and the regulation of blood vessel tone-diameter, which regulate the blood pressure and the blood flow, respectively. Under diabetic conditions, an increased flux of glucose through the HBP has been proposed to cause vascular disease. It has been observed that prolonged exposure to high glucose leads to the increase of GFAT expression in VSMCs [27], indicating that GFAT is possibly involved in the development of the diabetic vascular complications. Inhibition of GFAT activity using 6-diazo-5-oxonorleucine decreases the hyperglycemia-induced tumor growth factor-alpha (TGF-α) expression in VSMCs, suggesting that the adverse effects of hyperglycemia in VSMCs are mediated by the HBP. Hall et al. [28] demonstrated that expressions of GLUT1 and GLUT4 are increased in the neointima of the aorta after balloon injury. Increased proliferation and decreased apoptosis of VSMCs provides a possible linkage with the increased risks of restenosis and atherosclerosis in patients with diabetes. Akimoto et al. [29] found that the pattern of O-GlcNAc modification of proteins changed when rat aortic smooth muscle cells (RASMCs) were cultured in medium containing a high concentration of glucose. High glucose also elevates both the expression and activity of OGT [29]. In addition, high glucose and glucosamine also induced an increase in the expression of growth factors in RASMCs [30]. The effects of O-GlcNAc on cell growth and division may also contribute to the increase in VSMC proliferation seen in diabetes [31]. Treatment of VSMCs with high glucose increases the expressions of TGF-α and basic fibroblast growth factor, leading to abnormal proliferation of smooth muscle cells, a characteristic associated with atherosclerotic lesions [30]. It has been reported that an elevated glucose concentration in VSMCs causes an increase in the activity of protein kinase C (PKC), known to modulate the expression of several growth factors including TGF-α [32]. McClain et al. [30] also found that treatment of cells with phorbol ester to repress PKC and treatment of staurosporine to inhibit PKC did not affect the ability of glucose or glucosamine to activate the TGF-α promoter. Hattori et al. [33] found that hyperglycemia-induced activation of PKC was involved in the activation of TNFα-induced NF-κB, a transcription factor involved in the pathogenesis of atherosclerosis. Recently, Yang et al. [34] demonstrated that Thr352 of the P65 subunit of NF-κB was O-GlcNAcylated, which results in increases in its transcriptional activity and half-life within the nuclei of VSMCs cultured under high glucose conditions. They also found that attenuation of O-GlcNAcylation by over-expression of OGA inhibits the high glucose-induced NF-κB activation in VSMCs. On the other hand, over-expression of OGT along with OGA inhibitor increases NF-κB transcriptional activity. These results suggest that O-GlcNAc modification of transcription factors presumably modulates their activities.
- As described above, excess glucose flux through the HBP results in elevated UDP-GlcNAc levels in VSMCs, which may have the capacity to modify and regulate vascular reactivity. Proteins with an important role in vascular function, such as endothelial nitric oxide synthase (eNOS), sarcoplasmic reticulum Ca2+-ATPase, phospholipase C, PKC, and phosphoinositide-3-kinase, are also targets for O-GlcNAcylation, suggesting that O-GlcNAcylation may play a critical role in vascular reactivity [35]. In isolated rat thoracic aortic rings, increasing O-GlcNAc formation via PUGNAC blunted vascular relaxation by acetylcholine and augmented vasoconstriction in response to phenyleprine [36]. In addition, treatment with ST060266, an OGT inhibitor, inhibits the U46619-induced blood vessel contraction which depends on Rho kinase (RhoK) activity, suggesting that RhoK is also modified by GlcNAcylation (Kim DH and Jeoung NH, unpublished data). The contractile response of vascular smooth muscle is mediated through myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP) activations, which are initiated by the Ca2+-calmodulin interaction that causes phosphorylation of the myosin light chain (MLC) [37]. Clark et al. [38] demonstrated impaired calcium cycling via transcriptional inhibition of sarcoplasmic Ca2+-ATPase in OGT-over-expressing cardiomyocytes. It was demonstrated that membrane depolarization promotes OGT activation, leading to induction of O-GlcNAcylation of total proteins, which can be reduced by inhibition of either voltage-gated Ca2+ channels or Ca2+-calmodulin-dependent kinase IV (CAMIV), suggesting that OGT activity is regulated by Ca2+ influx and CAMIV-dependent phosphorylation. Interestingly, CAMIV has been shown to phosphorylate and activate OGT both in vivo and in vitro [39]. Smooth muscle cells also contain Ca2+-independent mechanisms to regulate contractibility. Removal of the phosphate group by MLCP results in inactivation of MLC and promotion of smooth muscle relaxation. MLCP consists of two subunits; one catalytic subunit of type 1 protein phosphatase (PP1cδ) and a non-catalytic subunit [40]. Recently Cheung et al. [41] found that MYPT1, a known targeting regulatory subunit of PP1cδ, is O-GlcNAcylated and also acts as a substrate for OGT. The substrate specificity of OGT is also regulated by MYPT1, which dephosphorylates OGT under different conditions. Based on the above findings, it is clear that O-GlcNAcylation on specific vascular proteins has an important role in the regulation vascular reactivity, and further research is necessary to determine the impact of O-GlcNAcylation on vascular reactivity.
O-GLCNACYLATION ON VASCULAR SMOOTH MUSCLE CELLS
- Endothelial cell dysfunction has emerged as a key component in the pathophysiology of cardiovascular abnormalities associated with diabetes [42]. Normal endothelial production of nitric oxide by eNOS plays a pivotal role in the regulation of vascular tone and remodeling, the inhibition of platelet aggregation and adhesion to the vascular wall, as well as the synthesis and secretion of extracellular matrix proteins, and proliferation of VSMCs [43]. Endothelial cells are sensitive to hyperglycemia because of their poor ability to regulate intracellular glucose [44]. In cultured bovine aortic endothelial cells, hyperglycemia caused an increase in mitochondrial superoxide production in association with an increase in O-GlcNAc on eNOS and a reciprocal decrease in phosphorylation at Ser1177, the site responsible for activation of the enzyme [45]. Inhibition of GFAT by antisense oligonucleotides blocks the O-GlcNAcylation of eNOS, confirming the role of increased HBP flux of glucose. This O-GlcNAc-induced decrease in eNOS activity was associated with increases in matrix metalloproteinase (MMP) activity and expression combined with decreased tissue inhibition of metalloproteinase (TIMP) expression [46]. An imbalance between MMPs and TIMPs has been implicated in atherosclerosis-related complications in diabetes [46]. Du et al. [47] demonstrated that hyperglycemia-induced expression of plasminogen activator inhibitor-1 (PAI-1) in endothelial cells was associated with an increase in O-GlcNAc levels on Sp1, which increases expression of TGF-1β, a potent inducer of extracellular matrix protein synthesis, leading to proliferation of VSMCs and fibroblasts. High glucose also induces O-GlcNAc modification of Sp3 and up-regulates angiopoietin-2 gene expression in the microvascular system [48]. This leads to the induction of ICAM-1 and VCAM-1 expressions and sensitization of microvascular endothelial cells to the proinflammatory effects of TNFα. Increased O-GlcNAcylation of Sp1 and Sp3 also regulates the endothelial cell-specific expression of vascular endothelium growth factor receptor [49]. These observations suggest that chronically elevated O-GlcNAc level represents a common mechanism underlying the adverse effects of hyperglycemia. The vascular effects of HBP/O-GlcNAc signaling are just beginning to be understood, and a more direct link between increased O-GlcNAc levels and vascular dysfunction remains to be discovered (Fig. 2).
O-GLCNACYLATION ON ENDOTHELIAL DYSFUNCTION
- Many studies have shown that a loss of control of the dynamics of O-GlcNAcylation interferes with the normal progress of fundamental events and may be implicated in the appearance of pathological states. However, since OGT is essential for cell viability and is highly conserved from an evolutionary perspective, its presence has to convey some survival advantage to cells and organisms [12]. It has been supported by a number of studies demonstrating that an acute increase in O-GlcNAc synthesis improves tolerance to a variety of stressful stimuli and increases cell survival [50,51]. When the O-GlcNAc response was inhibited by decreased OGT expression, cell viability also decreased, whereas augmentation of O-GlcNAc levels with PUGNAC increased cell survival [52]. Glucose uptake in cells has been linked to the capacity of cells to respond and survive deleterious cellular conditions. Even though the sustained hyperglycemia in T2D is clearly associated with the adverse effects at the cellular level, the acute increases in circulating glucose level and of glucose utilization in tissues have beneficial effects on cell survival. It has been reported that increased glucose utilization protects against a wide range of injuries like trauma, sepsis, shock, cardiac surgery, and myocardial infarction [50]. During acute heat stress conditions, an increase in O-GlcNAc levels enhances cell survival, whereas inhibition of GFAT decreases O-GlcNAc levels in the cells, leading to an increase in cell death [51]. Increasing the O-GlcNAc level results in cells that are more thermo-tolerant, which results in an increase in the expressions of heat shock protein 40 (HSP40) and HSP70 induced by the O-GlcNAc signaling pathway [53].
- Acute activation of O-GlcNAc formation inhibits acute inflammatory and neointimal responses to endoluminal vascular injury in vivo [54]. Further, increases in O-GlcNAc-modified proteins decrease the expressions of inflammatory mediators and the infiltration of leukocytes at the injured endoluminal carotid artery. Since augmentation of O-GlcNAc levels attenuates the activation of inflammatory mediators in acute cardiovascular injury, it is possible that an increased O-GlcNAc level in diabetes may be beneficial for its ability to attenuate the proinflammatory response. Glucosamine treatment during resuscitation improved cardiac function by reducing the trauma-hemorrhage-induced ICAM-1 expression, NF-κB expression, and NF-κB DNA binding activity in the heart [55], suggesting that acutely increased O-GlcNAc level may reduce the NF-κB-mediated inflammation.
- In isolated neonatal cardiomyocytes, hypoxia-reperfusion causes a transient increase in O-GlcNAc level and improves cell viability. Treatment of neonatal cardiomyocytes with glucosamine, hyperglycemia, or PUGNAC significantly increases O-GlcNAc level, improves cell viability, and decreases necrosis and apoptosis [56]. Schaffer et al. [57] reported that high glucose significantly reduces hypoxia-induced apoptosis and necrosis in isolated cardiomyocytes via decreased Ca2+ overload. Interestingly, cardiac mitochondria isolated from PUGNAC-treated mice and also OGT-over-expressing neonatal rat cardiomyocytes are resistant to Ca2+ induced mitochondrial membrane transition pore (mPTP) formation, a critical step in the initiation of apoptosis and cell death [58]. Recently, Champattanachai et al. [59] found that glucosamine, OGT over-expression, and O-GlcNAcase inhibition protect neonatal cardiomyocytes from ischemia-reperfusion injury via an increased level of mitochondrial bcl-2, a key regulatory protein for cell survival. A glucosamine-induced increase in bcl-2 inhibits mPTP opening via direct interaction with voltage-dependent anion channel 1α, one of the putative components of mPTP (Fig. 3).
- Thus, acute activation of metabolic pathways leading to an increase in O-GlcNAc level is an endogenous stress-activated response, and augmentation of this response improves short term protection in the cardiovascular system. Induction of prosurvival factors such as HSP or attenuation of the inflammatory response improves long term cardiovascular protection. Further studies on the function of O-GlcNAc-modified proteins in complex signaling networks will provide answers surrounding cell survival in response to stress.
O-GLCNACYLATION AND CARDIOVASCULAR PROTECTION
- In this review, we summarized the data supporting the beneficial effects related to acute increases in O-GlcNAc level, as well as the adverse effects produced by chronic activation. This suggests that the initial response to an acute stress induces protective processes and improves survival; as the load increases, the continued activation of the pathway results in the development of pathophysiology. Thus, altering the O-GlcNAc levels in vascular tissues may represent a novel therapeutic approach for the treatment of diabetic cardiovascular disease.
CONCLUSIONS
- 1. Copeland RJ, Bullen JW, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab 2008;295:E17-E28. ArticlePubMedPMC
- 2. Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science 2001;291:2376-2378. ArticlePubMed
- 3. Guinez C, Morelle W, Michalski JC, Lefebvre T. O-GlcNAc glycosylation: a signal for the nuclear transport of cytosolic proteins? Int J Biochem Cell Biol 2005;37:765-774. ArticlePubMed
- 4. Zachara NE, Hart GW. O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim Biophys Acta 2004;1673:13-28. ArticlePubMed
- 5. Butkinaree C, Park K, Hart GW. O-linked β-N-acetyl glucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta 2010;1800:96-106. ArticlePubMed
- 6. Bouche C, Serdy S, Kahn CR, Goldfine AB. The cellular fate of glucose and its relevance in type-2 diabetes. Endocr Rev 2004;25:807-830. ArticlePubMedPDF
- 7. Hanover JA. Glycan-dependent signaling: O-linked N-acetyl glucosamine. FASEB J 2001;15:1865-1876. ArticlePubMedPDF
- 8. Buse MG. Hexosamines, insulin resistance, and the complications of diabetes; current status. Am J Physiol Endocrinol Metab 2006;290:E1-E8. ArticlePubMed
- 9. Kornfeld R. Studies on L-glutamine D-fructose 6-phosphate amidotransferase. I. Feedback inhibition by uridine diphosphate-N-acetylglucosamine. J Biol Chem 1967;242:3135-3141. PubMed
- 10. Marshall S, Nadeau O, Yamasaki K. Dynamic actions of glucose and glucosamine on Hexosamine biosynthesis in isolated adipocytes: differential effects on glucosamine 6-phosphate, UDP-N-acetyl glucosamine and ATP levels. J Biol Chem 2004;279:35313-35319. PubMed
- 11. Haltiwanger RS, Holt GD, Hart GW. Enzymatic addition of O-GlcNAc to nuclear and cytosolic proteins. Identification of a uridine diphospho-N-acetyl glucosamine: peptide beta-Nacetylglucosaminyl transferase. J Biol Chem 1990;265:2563-2568. PubMed
- 12. Shafi R, Lyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X-chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A 2000;97:5735-5739. ArticlePubMedPMC
- 13. Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A 2008;105:13793-13798. ArticlePubMedPMC
- 14. Defronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia and atherosclerotic cardiovascular disease. Diabetes care 1991;14:173-194. ArticlePubMedPDF
- 15. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of Hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem 1991;266:4706-4712. ArticlePubMed
- 16. Marshall S. Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional prospective of diabetes, obesity and cancer. Sci STKE 2006;2006:re7PubMed
- 17. Rajapakse AG, Ming XF, Carvas JM, Yang Z. The hexosamine biosynthesis inhibitor azaserine prevents endothelial inflammation and dysfunction under hyperglycemic condition through antioxidant effects. Am J Physiol Heart Circ Physiol 2009;296:H815-H822. ArticlePubMed
- 18. Werstuck GH, Khan MI, Femia G, Kim AJ, Tedesco V, Trigatti B, Shi Y. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes 2006;55:93-101. ArticlePubMedPDF
- 19. Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in AKT activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A 2002;99:5313-5318. ArticlePubMedPMC
- 20. Cooksey RC, Hebert LF Jr, Zhu JH, Wofford P, Garrey WT, McClain DA. Mechanism of Hexosamine-induced insulin resistance in transgenic mice over-expressing glutamine: fructose-6-phosphate amidotransferase: decreased glucose transporter GLUT4 translocation and reversal by treatment with thiazolidinedione. Endocrinology 1999;140:1151-1157. ArticlePubMed
- 21. Brozinick JT Jr, Berkemeier BA, Elmendorf JS. Acting on GLUT4: membrane and cytoskeletal components of insulin action. Curr Diabetes Rev 2007;3:111-122. PubMedPMC
- 22. Bhonagiri P, Pattar GR, Horvath EM, Habegger KM, McCarthy AM, Elmendof JS. Hexosamine biosynthesis pathway flux contributes to insulin resistance via altering membrane phosphatidylinositol 4, 5-bisphosphate and cortical filamentous actin. Endocrinology 2009;150:1636-1645. ArticlePubMed
- 23. Gandy JC, Rountree AE, Bijur GN. Akt is dynamically modified with O-GlcNAc following treatments with PUGNAC and insulin-like growth factor-1. FEBS Lett 2006;580:3051-3058. PubMedPMC
- 24. Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, Kudlow JE, Michell RH, Olefsky JM, Field SJ, Evans RM. Phosphoinositide signaling links O-GlcNAc transferase to insulin resistance. Nature 2008;451:964-969. ArticlePubMedPDF
- 25. Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci 2005;109:143-159. ArticlePDF
- 26. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szado C, Brownlee M. Inhibition of GAPDH activity by poly (ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 2003;112:1049-1057. ArticlePubMedPMC
- 27. Nerlich AG, Sauer U, Kolm-Lilly V, Wagner E, Koch M, Schleicher ED. Expression of glutamine: fructose 6-phosphate amidotransferase in human tissues; evidence for high variability and distinct regulation in diabetes. Diabetes 1998;47:170-178. ArticlePubMed
- 28. Hall JL, Chatham JC, Eldar-Finkelman H, Gibbons GH. Upregulation of glucose metabolism during intimal lesion formation is coupled to the inhibition of vascular smooth muscle cell apoptosis: role of GSK3beta. Diabetes 2001;50:1171-1179. PubMed
- 29. Akimoto Y, Kreppel LK, Hirano H, Hart GW. Hyperglycemia and the O-GlcNAc transferase in rat aortic smooth muscle cells; elevated expression and altered patterns of O-GlcNAcylation. Arch Biochem Biophys 2001;389:166-175. ArticlePubMed
- 30. McClain DA, Paterson AJ, Roos MD, Wei X, Kudlow JE. Glucose and glucosamine regulate growth factor gene expression in vascular smooth muscle cells. Proc Natl Acad Sci U S A 1992;89:8150-8154. ArticlePubMedPMC
- 31. Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW. Perturbations in O-linked beta-N-acetyl glucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem 2005;280:32944-32956. PubMed
- 32. Williams B, Schrier RW. Characterization of glucose-induced in situ protein kinase C activity in cultured vascular smooth muscle cells. Diabetes 1992;41:1464-1472. ArticlePubMed
- 33. Hattori Y, Hattori S, Sato N, Kasai K. High-glucose-induced nuclear factor kB activation in vascular smooth muscle cells. Cardiovasc Res 2000;46:188-197. PubMed
- 34. Yang WH, Park SY, Nam HW, Kim do H, Kang JG, Kang ES, Kim YS, Lee HC, Kim KS, Cho JW. NF-kB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci U S A 2008;105:17345-17350. PubMedPMC
- 35. Fulop N, Marchase RB, Chatham JC. Role of protein O-linked N-acetyl glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res 2007;73:288-297. ArticlePubMed
- 36. Lima VV, Giachini FR, Carneiro FS, Carneiro ZN, Saleh MA, Pollock DM, Fortes ZB, Carvalho MH, Ergul A, Webb RC, Tostes RC. O-GlcNAcylation contributes to augmented vascular reactivity induced by endothelin 1. Hypertension 2010;55:180-188. ArticlePubMed
- 37. Hilgers RH, Webb RC. Molecular aspects of arterial smooth muscle contraction: focus on Rho. Exp Biol Med (Maywood) 2005;230:829-835. ArticlePubMedPDF
- 38. Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem 2003;278:44230-44237. ArticlePubMed
- 39. Song M, Kim HS, Park JM, Kim SH, Kim IH, Ryu SH, Suh PG. O-GlcNAc transferase is activated by CAMKIV-dependent phosphorylation under potassium chloride-induced depolarization in NG-108-15 cells. Cell Signal 2008;20:94-104. ArticlePubMed
- 40. Hirano K. Current topics in the regulatory mechanism underlying the Ca2+ sensitization of the contractile apparatus in vascular smooth muscle. J Pharmacol Sci 2007;104:109-115. ArticlePubMed
- 41. Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW. O-linked Beta-N-acetyl glucosaminyl transferease substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J Biol Chem 2008;283:33935-33941. PubMedPMC
- 42. Calles-Escandon J, Cipolla M. Diabetes and endothelial dysfunction: a clinical perspective. Endocr Rev 2001;22:36-52. ArticlePubMed
- 43. Makimattila S, Virkamaki A, Groop PH, Cockcroft J, Utriainen T, Fagerudd J, Yki-Jarvinen H. Chronic hyperglycemia impairs endothelial function and insulin sensitivity via different mechanisms in insulin dependent diabetes mellitus. Circulation 1996;94:1276-1282. ArticlePubMed
- 44. Yetik-Anacak G, Catravas JD. Nitric oxide and the endothelium: history and impact on cardiovascular disease. Vascul Pharmacol 2006;45:268-276. ArticlePubMed
- 45. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by post-translational modification at the AKT site. J Clin Invest 2001;108:1341-1348. ArticlePubMedPMC
- 46. Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 2002;106:466-472. ArticlePubMed
- 47. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A 2000;97:12222-12226. ArticlePubMedPMC
- 48. Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, Suske G, Rabbani N, Thornalley PJ, Sarthy VP, Hammes HP, Brownlee M. High glucose increases angiopoietin-2 transcription in micro vascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem 2007;282:31038-31045. ArticlePubMed
- 49. Hata Y, Duh E, Zhang K, Robinson GS, Aiello LP. Transcription factors Sp1 and Sp3 alter vascular endothelial growth factor receptor expression through a novel recognition sequence. J Biol Chem 1998;273:19294-19303. ArticlePubMed
- 50. Fath-Ordoubadi F, Beatt KJ. Glucose-insulin-potassium therapy for treatment of acute myocardial infarction: an overview of randomized placebo-controlled trials. Circulation 1997;96:1152-1156. ArticlePubMed
- 51. Sohn KC, Lee KY, Park JE, Do SI. OGT functions as a catalytic chaperone under heat stress response: a unique defense role of OGT in hyperthermia. Biochem Biophys Res Commun 2004;322:1045-1051. ArticlePubMed
- 52. Zachara NE, O'Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem 2004;279:30133-30142. PubMed
- 53. Lim KH, Chang HI. O-linked N-acetyl glucosamine suppresses thermal aggregation of Sp1. FEBS Lett 2006;580:4645-4652. ArticlePubMed
- 54. Xing D, Feng W, Not LG, Miller AP, Zhang Y, Chen YF, Majid-Hassan E, Chatham JC, Oparil S. Increased protein O-GlcNAc modification inhibits inflammatory and neointimal responses to acute endoluinal arterial injury. Am J Physiol Heart Circ Physiol 2008;295:H335-H342. PubMedPMC
- 55. Zou L, Yang S, Champattanachai V, Shunhua HU, Chaudry IH, Marchase RB, Chatham JC. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-kB signaling. Am J Physiol Heart Circ Physiol 2009;296:H515-H523. PubMed
- 56. Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein-associated O-GlcNAc. Am J Physiol Cell Physiol 2007;292:C178-C187. ArticlePubMed
- 57. Schaffer SW, Croft CB, Solodushko V. Cardioprotective effect of chronic hyperglycemia: effect on hypoxia-induced apoptosis and necrosis. Am J Physiol Heart Circ Physiol 2000;278:H1948-H1954. ArticlePubMed
- 58. Ngoh GA, Jones SP. O-GlcNAc signaling attenuates mitochondrial membrane permeability transition. FASEB J 2008 22:1130.8.[Meeting Abstract].
- 59. Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am J Physiol Cell Physiol 2008;294:C1509-C1520. ArticlePubMed
REFERENCES
Fig. 1Interplay between O-GlcNAcylation and protein phosphorylation. (A) O-GlcNAc modification is strongly dependent on the concentration of UDP-GlcNAc produced by the hexosamine biosynthetic pathway (HBP). OGT uses UDP-GlcNAc as a substrate for GlcNAcylation on protein serine and threonine residues. O-GlcNAcase (OGA) removes the O-GlcNAc moiety from O-GlcNAc-modified proteins. PUGNAC inhibits the activity of O-GlcNAcase. (B) O-GlcNAc modification is analogous to protein phosphorylation/dephosphorylation. Interplay between GlcNAcylation and phosphorylation can affect the activities or stabilities of the proteins, and both processes can occur on the same protein at proximal sites.
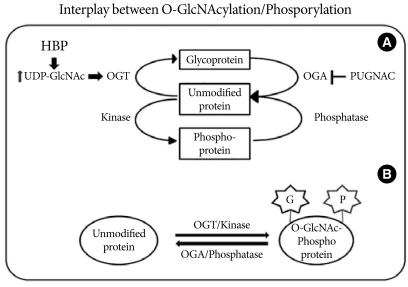
Fig. 2A fraction (2-5%) of the glucose entering a cell is directed into the hexosamine biosynthesis pathway (HBP) pathway. GFAT (glutamine: fructose-6-phosphate amidotransferase) uses glutamine to convert fructose-6-phosphate into glucosamine-6-phosphate, which is then used for the synthesis of UDP-GlcNAc in the cell. Activation of the HBP pathway acts through Sp1 sites to increase plasminogen activator inhibitor-1 (PAI-1) and tumor growth factor-alpha expressions. At the same time, O-GlcNAc modification of endothelial nitric oxide synthase (eNOS) decreases its activity and nitric oxide (NO) production.
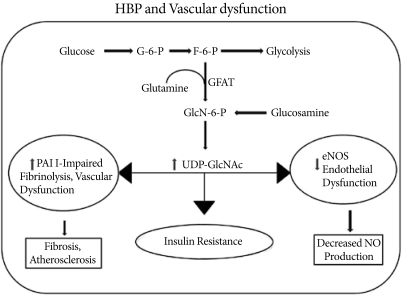
Fig. 3Hyperglycemia-induced activation of O-linked N-acetyl glucosamine activates GPX1 and its binding to c-Abl and Arg kinases and protects the cell via the antioxidant response. Along with an increase in the antioxidant response, increased O-GlcNAcylation activates p38 MAPK phosphorylation with increased glucose transport. Increased O-GlcNAc formation also increases the level of mitochondrial Bcl2, which inhibits apoptosis and protects the cell during stress.
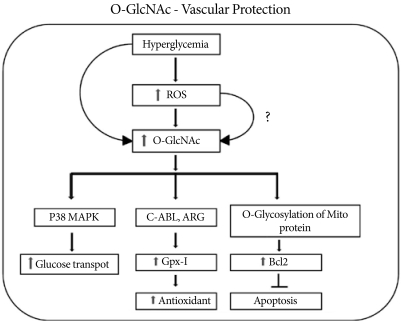
Figure & Data
References
Citations
Citations to this article as recorded by 
- Human Pulmonary Artery Endothelial Cells Increased Glycolysis and Decreased Nitric Oxide Synthase O-GlcNAcylation in Pulmonary Arterial Hypertension
Sarah E. Basehore, Alisa Morss Clyne
International Journal of Translational Medicine.2024; 4(1): 140. CrossRef - Hyperglycemia Aggravates the Cerebral Ischemia Injury via Protein O-GlcNAcylation
Jing Zhu, Xin Ji, Ruirui Shi, Tianqi He, Su-ying Chen, Ruochen Cong, Bosheng He, Su Liu, Hui Xu, Jin-hua Gu, Chunling Dai
Journal of Alzheimer's Disease.2023; 94(2): 651. CrossRef - Hyper-O-GlcNAcylation impairs insulin response against reperfusion-induced myocardial injury and arrhythmias in obesity
Lingyan Jin, Feng Gao, Tiannan Jiang, Binghua Liu, Caiyao Li, Xinghua Qin, Qiangsun Zheng
Biochemical and Biophysical Research Communications.2021; 558: 126. CrossRef - Laminar Flow on Endothelial Cells Suppresses eNOS O-GlcNAcylation to Promote eNOS Activity
Sarah E. Basehore, Samantha Bohlman, Callie Weber, Swathi Swaminathan, Yuji Zhang, Cholsoon Jang, Zoltan Arany, Alisa Morss Clyne
Circulation Research.2021; 129(11): 1054. CrossRef - OGT knockdown counteracts high phosphate-induced vascular calcification in chronic kidney disease through autophagy activation by downregulating YAP
Tian-Hua Xu, Zitong Sheng, Yue Li, Xiaobo Qiu, Binyao Tian, Li Yao
Life Sciences.2020; 261: 118121. CrossRef - OGT-Mediated KEAP1 Glycosylation Accelerates NRF2 Degradation Leading to High Phosphate-Induced Vascular Calcification in Chronic Kidney Disease
Tian-Hua Xu, Yinke Du, Zitong Sheng, Yue Li, Xiaobo Qiu, Binyao Tian, Li Yao
Frontiers in Physiology.2020;[Epub] CrossRef - The Role of Stress-Induced O-GlcNAc Protein Modification in the Regulation of Membrane Transport
Viktória Fisi, Attila Miseta, Tamás Nagy
Oxidative Medicine and Cellular Longevity.2017; 2017: 1. CrossRef - Oral administration of probiotic Lactobacillus paraplantarum BGCG11 attenuates diabetes-induced liver and kidney damage in rats
Mirjana Mihailović, Milica Živković, Jelena Arambašić Jovanović, Maja Tolinački, Marija Sinadinović, Jovana Rajić, Aleksandra Uskoković, Svetlana Dinić, Nevena Grdović, Nataša Golić, Melita Vidaković
Journal of Functional Foods.2017; 38: 427. CrossRef - Cardiovascular Protection by Sodium Glucose Cotransporter 2 Inhibitors: Potential Mechanisms
Bart Staels
The American Journal of Cardiology.2017; 120(1): S28. CrossRef - Stable Isotope Labeling with Amino Acids (SILAC)-Based Proteomics of Primary Human Kidney Cells Reveals a Novel Link between Male Sex Hormones and Impaired Energy Metabolism in Diabetic Kidney Disease
Sergi Clotet, Maria Jose Soler, Marta Riera, Julio Pascual, Fei Fang, Joyce Zhou, Ihor Batruch, Stella K. Vasiliou, Apostolos Dimitromanolakis, Clara Barrios, Eleftherios P. Diamandis, James W. Scholey, Ana Konvalinka
Molecular & Cellular Proteomics.2017; 16(3): 368. CrossRef - Cardiovascular Protection by Sodium Glucose Cotransporter 2 Inhibitors: Potential Mechanisms
Bart Staels
The American Journal of Medicine.2017; 130(6): S30. CrossRef - Nutrient regulation of transcription and signalling by O-GlcNAcylation
Gerald W. Hart
Perspectives in Science.2015; 6: 49. CrossRef - β-Glucan administration to diabetic rats alleviates oxidative stress by lowering hyperglycaemia, decreasing non-enzymatic glycation and protein O-GlcNAcylation
Mirjana Mihailović, Jelena Arambašić, Aleksandra Uskoković, Svetlana Dinić, Nevena Grdović, Jelena Marković, Jelena Bauder, Goran Poznanović, Melita Vidaković
Journal of Functional Foods.2013; 5(3): 1226. CrossRef - DecreasedO-GlcNAcylation of the key proteins in kinase and redox signalling pathways is a novel mechanism of the beneficial effect of α-lipoic acid in diabetic liver
Svetlana Dinić, Jelena Arambašić, Mirjana Mihailović, Aleksandra Uskoković, Nevena Grdović, Jelena Marković, Borivoje Karadžić, Goran Poznanović, Melita Vidaković
British Journal of Nutrition.2013; 110(3): 401. CrossRef - Modulation of Dynamin-related Protein 1 (DRP1) Function by Increased O-linked-β-N-acetylglucosamine Modification (O-GlcNAc) in Cardiac Myocytes
Thomas Gawlowski, Jorge Suarez, Brian Scott, Moises Torres-Gonzalez, Hong Wang, Raphaela Schwappacher, Xuemei Han, John R. Yates, Masahiko Hoshijima, Wolfgang Dillmann
Journal of Biological Chemistry.2012; 287(35): 30024. CrossRef -
O-GlcNAcylation Increases ChREBP Protein Content and Transcriptional Activity in the Liver
Céline Guinez, Gaëlle Filhoulaud, Fadila Rayah-Benhamed, Solenne Marmier, Céline Dubuquoy, Renaud Dentin, Marthe Moldes, Anne-Françoise Burnol, Xiaoyong Yang, Tony Lefebvre, Jean Girard, Catherine Postic
Diabetes.2011; 60(5): 1399. CrossRef
 PubReader
PubReader Cite
Cite