The Effects of Glyburide on Apoptosis and Endoplasmic Reticulum Stress in INS-1 Cells in a Glucolipotoxic Condition
Article information
Abstract
Background
β-cell death due to endoplasmic reticulum (ER) stress has been regarded as an important pathogenic component of type 2 diabetes. The possibility has been suggested that sulfonylurea, currently being used as one of the main oral hypoglycemic agents of type 2 diabetes, increases ER stress, which could lead to sulfonylurea failure. The authors of the present study examined ER stress of β-cells in a glucolipotoxic condition using glyburide (GB) in an environment mimicking type 2 diabetes.
Methods
Apoptosis was induced by adding various concentrations of GB (0.001 to 200 µM) to a glucolipotoxic condition using 33 mM glucose, and the effects of varied concentrations of palmitate were evaluated via annexin V staining. The markers of ER stress and pro-apoptotic markers were assessed by Western blotting and semi-quantitative reverse transcription-polymerase chain reaction. Additionally, the anti-apoptotic markers were evaluated.
Results
Addition of any concentration of GB in 150 µM palmitate and 33 mM glucose did not increase apoptosis. The expression of phosphorylated eukaryotic initiation factor (eIF-2α) was increased and cleaved caspase 3 was decreased by adding GB to a glucolipotoxic condition. However, other ER stress-associated markers such as Bip-1, X-box binding protein-1, ATF-4 and C/EBP-homologous protein transcription factor and anti-apoptotic markers phosphor-p85 phosphatidylinositol 3-kinase and phosphorylation of Akt did not change significantly.
Conclusion
GB did not show further deleterious effects on the degree of apoptosis or ER stress of INS-1 cells in a glucolipotoxic condition. Increased phosphorylation of eIF-2α may attenuate ER stress for adaptation to increased ER protein load.
INTRODUCTION
The incidence of type 2 diabetes has sharply increased. Type 2 diabetes is characterized by insulin resistance and progressive decline in β-cell function. When β-cells do not compensate for insulin resistance and increased apoptosis, type 2 diabetes can develop [1]. Decreased β-cell function and mass are key factors in type 2 diabetes. Persistent hyperglycemia and elevated free fatty acids are suggested as a cause of β-cell failure and can occur via numerous mechanisms, including reactive oxygen species (ROS), increased intracellular calcium and the activation of endoplasmic reticulum (ER) stress. These processes have detrimental effects on β-cells by impairing insulin secretion, decreasing insulin gene expression and inducing apoptosis [2,3].
Pancreatic β-cells regulate insulin production and secretion to control blood glucose levels. In hyperglycemia, β-cells secrete insulin, which activates proinsulin biosynthesis in the ER of β-cells [4]. Therefore, β-cells are highly specialized to handle the protein load within the ER. ER homeostasis, the dynamic balance between the ER protein load and the ER capacity to process this load, is important for proper protein folding. Disruption of ER homeostasis leads to accumulation of unfolded and misfolded proteins in the ER. This condition is referred to as ER stress [5,6] and has been postulated to result from increased biosynthetic demand induced by chronic hyperglycemia and elevated free fatty acids in the β-cells. This pathway is well understood in the context of the unfolded protein response (UPR), which relieves ER stress, restores homeostasis, and prevents cell death by inducing numerous downstream responses that decrease new protein arrival to the ER, increase the amount of ER chaperones to improve folding capacity, and increase a cell's capacity to eliminate misfolded proteins. If unable to successfully perform these tasks, the UPR will trigger the apoptosis cascade [7]. The three primary modulators of the UPR are inositol requiring protein 1-α (IRE1-α), activating transcription factor 6 (ATF6), and protein kinase RNA (PRK)-like ER associated kinase (PERK) [8]. These sensors remain inactive via interaction with the ER chaperone BiP until activated by increased ER stress [9].
Sulfonylurea drugs, which reduce blood glucose levels by stimulating insulin release from pancreatic β-cells [10], have been used in the treatment of type 2 diabetes since the early 1950s. Despite the worldwide use of sulfonylureas, loss of β-cell mass and function has raised concern regarding the use of sulfonylureas for the treatment of type 2 diabetes mellitus. Studies have shown that sulfonylureas may induce apoptosis in β-cell lines and rodent islets [11], and sulfonylurea therapy failure is also very common in long-term treatment [12] though the mechanism is still unclear. However, some evidence has suggested that chronic use of sulfonylurea leads to ER stress in the β-cells, which finally causes exhaustion of β-cell function [13], and the decline in β-cell function causes the progressive deterioration of glycemic control. Qian et al. [14] suggested the hypothesis that sulfonylurea induces the loss of β-cell function and influences the natural history of the disease through acceleration of ER stress. The use of sulfonylureas for the treatment of type 2 diabetes mellitus may accelerate the loss of β-cell mass and function. Despite inconsistencies regarding types of sulfonylureas, previous results for these agents were generally negative [10-15]. A majority of the previous experiments showed conflicting results regarding sulfonylureas in β-cells without stresses or with only glucotoxicity, conditions which are quite different from the internal conditions of diabetic patients. Thus, we aimed to assess the degree of apoptosis and ER stress of INS-1 cells using glyburide (GB) in a glucolipotoxic condition mimicking diabetes.
METHODS
Cell culture
Rat insulinoma INS-1 cells were obtained from Yeungnam University in Korea and were maintained in RPMI1640 medium containing 10% fetal bovine serum (FBS), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 11 mM glucose, and 50 µM 2-mercaptoethanol. All experiments were incubated at 37℃ in 5% CO2 and were studied between the 30th and 40th passages.
Glucolipotoxic condition
INS-1 cells were plated in six-well plates at 5×104 cells per well at the appropriate conditions. The cells were incubated for 24 hours in various concentrations of palmitate (100 to 500 µM) with 33 mM glucose, typically used as a high glucose concentration [16]. To collect single cells, cells were treated with trypsin-ethylenediaminetetraacetic acid (EDTA) and centrifuged at 1,500 rpm for 5 minutes at 4℃. After aspirating the supernatants, cells were washed with 1 mL of annexin V binding solution (140 mM NaCl, 10 mM HEPES pH 7.4, 2.5 mM CaCl2) and centrifuged at 1,500 rpm for 5 minutes at 4℃. Supernatants were removed, and 3 µL of annexin V-fluorescein isothiocyanate (FITC) and 10 µL of propidium iodide were added. After incubation for 15 minutes in the dark, 300 µL of fluorescence activated cell sorting (FACS) buffer (1% FBS, 0.1% NaN3) was added, and the sample was analyzed by FACSort (BectonDickinson, BD Bioscience, San Jose, CA, USA). After five repetitions, the concentration of palmitate producing 30% to 50% apoptosis in the INS-1 cells was chosen as the glucolipotoxic condition.
GB effect on apoptosis in a glucolipotoxic condition
The procedures to evaluate apoptosis were the same as those for achieving the glucolipotoxic condition except for the incubating media. INS-1 cells plated in six-well plates were incubated in various concentrations of GB (0.001 to 200 µM) for 24 hours and then with medium containing 150 µM palmitate and 33 mM glucose for 24 hours. Apoptosis was assessed by annexin V staining and FACSort. The experiment was performed in triplicate.
Change in markers representing ER stress and the anti-apoptotic pathway by adding GB to a glucolipotoxic condition
The ER stress markers and anti-apoptotic defense were evaluated using INS-1 cells incubated with 10 and 100 µM GB in a glucolipotoxic condition via semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR) and Western blotting. The ER stress markers such as Bip-1, ATF-4, X-box binding protein-1 (XBP-1) and C/EBP-homologous protein transcription factor (CHOP) were assessed by RT-PCR. Phosphorylated eukaryotic initiation factor (eIF)-2α, caspase 3, and cleaved caspase 3 were evaluated by Western blotting. Additionally, the anti-apoptotic markers phosphor-p85 phosphatidylinositol 3-kinase (PI3K) and phosphorylation of Akt were also appraised by Western blotting. The experiment was performed in triplicate.
Reverse transcription polymerase chain reaction (RT-PCR)
Total cellular RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized by PCR using primers of ER stress markers from a premix RT-PCR kit (Bioneer, Daejeon, Korea). The following primer sequences were used: ATF-4; forward, 5'-TCTGTATGAGCCCTGAGTCCTACCT-3'; reverse, 5'-GGTCATAAGGTTTGGGTCGAGAACCAC-3', Bip-1; forward, 5'-GAGATTGTTCTGGTTGGCGGATCTACTC-3'; reverse, 5'-CCATATGCTACAGCCTCATCTGGGTT-3', CHOP; forward, 5'-CCTGAAAGCAGAAACCGGTC-3'; reverse, 5'-CCTCATACCAGGCTTCCAGC-3', XBP-1; forward, 5'-AAACAGAGTAGCAGCACAGACTGC-3'; reverse, 5'-GGATCTCTAAGACTAGAGGCTTGGTG-3', and GAPDH; forward, 5'-TCCCTCAAGATTGTCAGCAA-3'; reverse, 5'-AGATCCACAACGGATACATT-3'. Amplification was performed under the following conditions using a MyCycler thermal cycler (Bio-Rad, Hercules, CA, USA): pre-denaturation at 95℃ for 2 minutes; denaturation at 95℃ for 30 seconds annealing at 40℃ for 30 seconds; extension at 72℃ for 30 seconds, and final extension at 72℃ for 7 minutes. After amplification, 5 µL of the PCR products were subjected to electrophoresis on 1.5% agarose gels. The gels were visualized by a SL-20 DNA Image Visualizer (Seoulin, Seoul, Korea).
Western blotting
INS-1 cells were washed with phosphate buffered saline (PBS) and lysed in mammalian tissue lysis/extraction reagent including protease inhibitors and sodium orthovanadate. Protein was quantified using the BCA protein assay kit with 1× sodium dodecyl sulfate (SDS) sample buffer (50 mM Tris pH 6.8, 2% SDS, 10% glycerol, 50 mM DTT, and 0.01% bromophenol blue). Proteins were separated via 12% SDS-polyacrylamide gel electrophoresis (PAGE), transferred onto a polyvinylidene fluoride (PVDF) membrane, and immunoblotted with anti-PI3K (Tyr 458) (1:1,000), anti-total Akt (1:1,000), anti-phospho Akt (Ser 473) (1:1,000), anti-caspase 3 (1:1,000), anti-eIF-2α (1:1,000), anti-phosphoserine 51 eIF-2α (1:1,000), and anti-β-actin (1:1,000) at 4℃ overnight. The secondary antibody goat anti-rabbit conjugated alkaline phosphatase was applied for 1 hour at room temperature, and the membrane was developed using an AP-conjugated development kit (Bio-Rad). Developed protein bands were quantified by the Multi Gauge V2.2 program.
RESULTS
Glucolipotoxic condition
Apoptosis of INS-1 cells cultured in 33 mM glucose and 100-500 µM concentrations of palmitate increased dose-dependently. The concentration of palmitate constantly achieving 30-50% apoptosis in repeated experiments was chosen as the glucolipotoxic condition. The medium containing 150 µM of palmitate resulted in 42.6±10.52% apoptosis (Fig. 1).
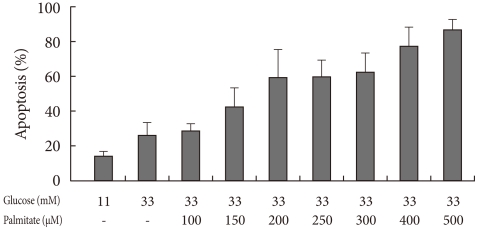
Apoptosis in INS-1 cells after incubation for 24 hours in culture media with 33 mM glucose and various concentrations of palmitate according to annexin V staining. Values are presented as mean±standard deviation of five repetitions.
GB effect on apoptosis in a glucolipotoxic condition
Addition of 0.001 to 200 µM GB in 150 µM palmitate and 33 mM glucose did not significantly induce apoptosis (Fig. 2). Rather, apoptosis tended to decrease in media containing 0.01 µM GB compared to that in the glucolipotoxic condition, although not significantly.
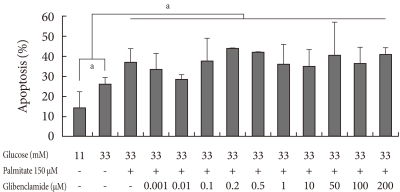
The effect on apoptosis of INS-1 cells at various concentrations of glyburide in a glucolipotoxic condition. Apoptosis was evaluated using annexin V staining. The experiments were performed in triplicate. Values are presented as mean±standard deviation and aP<0.05.
Changes in ER stress markers due to the addition of GB to a glucolipotoxic condition
Changes of the markers in the early cascade of ER stress according to GB addition to a glucolipotoxic condition
The ER stress markers Bip-1 (Fig. 3A), ATF-4 (Fig. 3B), XBP-1 (Fig. 3C), and phosphorylated eIF-2α (Fig. 3D) increased in glucotoxic and glucolipotoxic conditions compared to the levels observed in the normal glucose controls. The addition of GB into INS-1 cells in the glucolipotoxic condition did not significantly increase the expressions of these ER stress markers, and Bip-1 and ATF-4 tended to decrease with the addition of GB compared to the glucolipotoxic only condition, although the change was not significant. In addition, phosphorylation of eIF-2α showed an increase when GB was added to the glucolipotoxic condition. Conversely, according to the early cascade of markers of ER stress used in the present study, the phosphorylation of eIF-2α showed a significant reduction after GB addition compared to the levels in the glucolipotoxic condition.

Changes in endoplasmic reticulum stress markers with the addition of glyburide to a glucolipotoxic condition. Bip-1 (A), ATF-4 (B), and XBP-1 (C) were evaluated using reverse transcription-polymerase chain reaction. Phosphorylated eukaryotic initiation factor (eIF)-2α and eIF-2α were analyzed by Western blotting, and the phosphorylation rate was assessed (D). The experiments were performed in triplicate. Values are presented as mean±standard deviation and aP<0.05 for results under a glucolipotoxic condition.
Changes in ER stress markers representing the apoptotic pathway initiated by the addition of GB to a glucolipotoxic condition
Functional caspase 3 decreased and cleaved caspase 3 increased accordingly from control conditions to those of glucotoxicity and glucolipotoxicity (Fig. 4A and B). In other words, the cleaved caspase 3 form was increased compared to that of functional caspase 3. Although caspase 3 did not change due to the addition of GB to the glucolipotoxic condition, cleaved caspase 3 decreased significantly. Another pro-apoptotic marker CHOP was not significantly affected by the addition of GB (Fig. 4C).
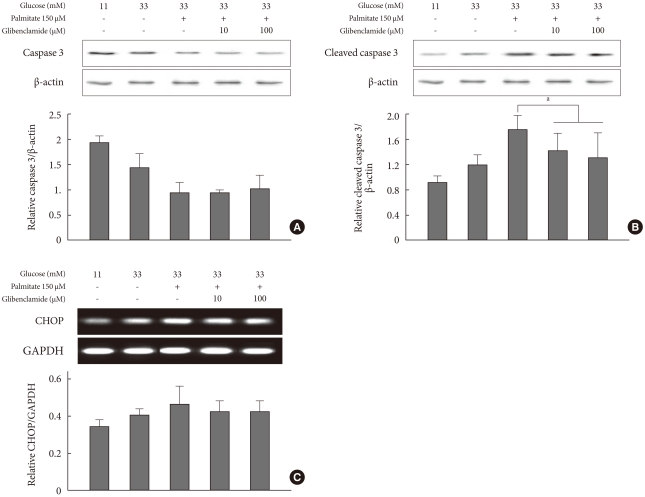
Changes in endoplasmic reticulum stress markers representing the pro-apoptotic pathway by adding glyburide to a glucolipotoxic condition. Functional caspase 3 (A) and cleaved caspase 3 (B) were evaluated according to Western blotting. CHOP (C) was assessed by reverse transcription-polymerase chain reaction. The experiments were performed in triplicate. Values are presented as mean±standard deviation and aP<0.05 for results under a glucolipotoxic condition.
Changes in anti-apoptotic markers
The markers representing apoptotic stress, PI3K (Fig. 5A) and phosphorylated Akt (Fig. 5B), tended to decrease in a β-cell damaged state such as glucotoxicity or glucolipotoxicity. However, the markers did not show any differences with the addition of GB to a glucolipotoxic condition.
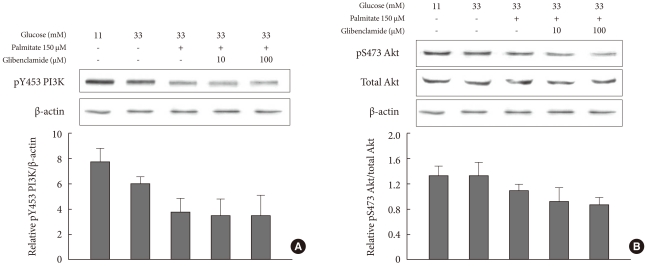
Changes in PI3K (A) and phosphorylated Akt (B), markers representing anti-apoptotic defense, due to the addition of glyburide to a glucolipotoxic condition. The experiments were performed in triplicate. Values are presented as mean±standard deviation.
DISCUSSION
The term glucolipotoxicity has emerged after recognition that the alterations in intracellular lipid partitioning underlying the mechanisms of lipotoxicity are dependent upon elevated glucose levels [17]. Prolonged exposure of isolated islets or insulin-secreting cells to elevated levels of fatty acids induces the inhibition of glucose-stimulated insulin secretion (GSIS) [18,19], impairment of insulin gene expression [20], and induction of cell death by apoptosis [16,21-23]. Evidence for ER stress in islets from type 2 diabetics has been shown through an increase in ER chaperones and CHOP along with enlarged ER [24-26].
Concern has been raised because studies have shown that sulfonylureas may induce β-cell apoptosis. In a recent study, β-cell apoptosis was induced by GB as well as the non-sulfonylurea secretagogues repaglinide and nateglinide in isolated human islets [15]. Glyburide of 0.1 and 10 µM induced 2.09- and 2.46-fold increases in β-cell apoptosis, whereas repaglinide did not change the number of apoptotic β-cells. At low concentration, nateglinide did not induce β-cell apoptosis, although a 1.49-fold increase in the number of apoptotic β-cells was observed at high concentrations. On the fourth day after exposure of the islets to secretagogues, β-cell apoptosis was apparent for all secretagogues.
Additionally, another study using human islets assessed the insulin content, GSIS, islet cell apoptosis, and mRNA expression of insulin and glucose transporter-1 in isolated human islets cultured in the presence of therapeutic concentrations of glimepiride (10 µmol/L), GB (10 µmol/L), or chlorpropamide (600 µmol/L) [10]. Insulin content decreased significantly after culture with all three sulfonylureas. Insulin responsiveness to glucose was preserved in islets incubated with glimepiride but not when islets were pre-incubated with GB or chlorpropamide.
Several studies have reported that chronic use of sulfonylureas increases the level of proinsulin (misfolded product of ER in β-cells) in the plasma of type 2 diabetes, indicating disequilibrium between ER load and folding capacity in β-cells [27,28]. Previous studies of β-cell apoptosis due to sulfonylurea were conducted at normal glucose concentrations or glucotoxic conditions in each cell line. However, the present study was conducted under a glucolipotoxic condition, which elevated ER stress, mimicking the internal environment of diabetic patients. Interestingly, apoptosis did not increase after addition of GB, a second-generation sulfonylurea, to create a glucolipotoxic condition (Fig. 2). Instead, apoptosis tended to decrease in media containing 0.01 µM GB compared to that in a glucolipotoxic condition, although not significantly. One probable hypothesis for this occurrence is the binary switch in ER stress [29]. The UPR regulates both adaptive and apoptotic effectors. The balance between the effectors depends on the nature of the ER stress, whether tolerable or intolerable. On sensing ER stress, IRE1 undergoes oligomerization and transautophosphorylation to activate its endoribonuclease domain. Activated IRE1 cleaves an intron from the mRNA encoding XBP-1. The spliced variant of XBP1 mRNA encodes a transcriptional activator for several UPR genes including chaperones, protein folding catalysts, and ER-associated degradation (ERAD) components. In addition to homeostatic functions, IRE1 also regulates apoptotic effectors. In the presence of unresolvable ER stress, IRE1 activates JNK through ASK1 and elicits apoptosis. This pathway has been shown to block the function of the anti-apoptotic Bcl-2 via phosphorylation, thus causing apoptosis in β-cells. IRE1 is also involved in the decay of the mRNAs encoding ER homeostatic proteins, including PDI, and BiP. Thus, IRE1 may be a major determinant of cell death. Under tolerable ER stress conditions, the UPR promotes β-cell survival. In contrast, under unresolvable ER stress conditions, the UPR induces β-cell death. Cells are exposed to physiological conditions that induce tolerable ER stress. Under ER stress conditions, the UPR can restore ER homeostasis to promote cell survival. Tight control of eIF2α phosphorylation is critical to ensure proper adaptation to increases in ER protein load and to promote β-cell survival [30-33]. Insulin biosynthesis stimulated by high glucose is markedly enhanced in PERK knockout mice as compared with that in control mice [30]. As a consequence, PERK knockout mice develop diabetes because of ER stress-mediated β-cell death. IRE1 is also activated under transient high glucose conditions. Acute IRE1 activation is required for proinsulin biosynthesis and perhaps enhancement of the ER proinsulin folding capacity [34]. The above-mentioned observations demonstrate that cells utilize the UPR in order to handle physiological disruptions of ER homeostasis to promote survival.
In the present study, most of the ER stress markers did not show significant differences after GB addition. Nevertheless, eIF2α phosphorylation was increased by GB, and proapoptotic CHOP tended to decrease, although not significantly. The data support the possibility that GB acts in agreement with an adaptive pathway of ER stress.
The United Kingdom Prospective Diabetes Study (UKPDS) determined that the loss of β-cell function was not unique to sulfonylureas but occurred at the same rate of decline in type 2 diabetic patients on metformin or those on conventional treatment [35]. This indicates that the use of sulfonylureas in type 2 diabetes may not be the direct cause of secondary β-cell failure.
The present study has several limitations regarding the cell line, type of sulfonylurea, and utilization of only in vitro data, as well as lack of a more detailed mechanism. Only a single β-cell line, INS-1, was used. To support the results, further studies will be needed using various cell lines and primary cell cultures of rodent and humans. Additionally, among the sulfonylureas, only GB was used. Several studies demonstrated that recently developed sulfonylureas did not increase apoptosis [10,36]. If GB does not induce apoptosis, other sulfonylureas currently being used and for which better data have been collected could be presumed to produce more favorable results; the authors intend to evaluate these recently developed sulfonylureas. Another limitation of the present study is the inclusion of only in vitro experiments. Although animal models of type 2 diabetes may represent internal glucolipotoxic conditions, the ability to measure the degree of glucolipotoxicity is difficult, and in vivo studies cannot rule out the possibility of interaction with another parameter. However, a well-designed in vivo experiment will be needed to confirm the results. In addition, the present study cannot explain a more detailed mechanism. Recently, several studies have reported that anti-apoptotic markers such as apoptosis antagonizing transcription factor (AATF) [37] and PI3K/Akt pathway [38] are associated with ER stress. In the present study, the induction of apoptosis by the addition of GB to a glucolipotixic condition did not show significant changes despite a decreasing tendency. Therefore, PI3K and Akt did not show direct correlation with an anti-apoptotic effect, although the pathway did not produce any harmful effects.
GB did not show further deleterious effects on the degree of apoptosis or ER stress of INS-1 cells in a glucolipotoxic condition. Increased phosphorylation of eIF-2α may attenuate ER stress for adaptation to increased ER protein load. The use of sulfonylurea in type 2 diabetes may not be the direct cause of secondary β-cell failure. To evaluate the results, further well-designed studies using various types of cell lines and sulfonylureas will be necessary to elucidate a more detailed mechanism.
Notes
No potential conflict of interest relevant to this article was reported.
ACKNOWLEDGMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009-0088556).