- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 45(5); 2021 > Article
-
ReviewTechnology/Device Assessment of Insulin Secretion and Insulin Resistance in Human
-
So Young Park1
 , Jean-François Gautier2,3, Suk Chon1,4
, Jean-François Gautier2,3, Suk Chon1,4 -
Diabetes & Metabolism Journal 2021;45(5):641-654.
DOI: https://doi.org/10.4093/dmj.2021.0220
Published online: September 30, 2021
1Department of Endocrinology and Metabolism, Kyung Hee University Hospital, Seoul, Korea
2Department of Diabetes, Clinical Investigation Centre (CIC-9504), Lariboisière Hospital, University Paris-Diderot, Paris, France
3Faculty of Medicine, University Paris-Diderot, Paris, France
4Department of Endocrinology and Metabolism, College of Medicine, Kyung Hee University, Seoul, Korea
-
Corresponding authors: Suk Chon, , Department of Endocrinology & Metabolism, College of Medicine, Kyung Hee University, 26 Kyunghee-dearo, Dongdaemungu, Seoul 02447, Korea E-mail: imdrjs@khu.ac.kr
-
Jean-François Gautier, , Department of Diabetes and Endocrinology, Lariboisière Hospital, University Paris 7, 2 Rue Ambroise Paré, Paris 75010, France E-mail: jean-francois.gautier@lrb.aphp.fr
Copyright © 2021 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The impaired insulin secretion and increased insulin resistance (or decreased insulin sensitivity) play a major role in the pathogenesis of all types of diabetes mellitus (DM). It is very important to assess the pancreatic β-cell function and insulin resistance/sensitivity to determine the type of DM and to plan an optimal management and prevention strategy for DM. So far, various methods and indices have been developed to assess the β-cell function and insulin resistance/sensitivity based on static, dynamic test and calculation of their results. In fact, since the metabolism of glucose and insulin is made through a complex process related with various stimuli in several tissues, it is difficult to fully reflect the real physiology. In order to solve the theoretical and practical difficulties, research on new index is still in progress. Also, it is important to select the appropriate method and index for the purpose of use and clinical situation. This review summarized a variety of traditional methods and indices to evaluate pancreatic β-cell function and insulin resistance/sensitivity and introduced novel indices.
- The prevalence of diabetes mellitus (DM) is increasing among people of all ages worldwide, becoming a major burden on public health care [1]. Insulin, a peptide hormone composed of 51 amino acids is the most important hormone for energy metabolism such as glucose, fat and protein and for maintaining homeostasis. The impaired insulin secretion and action are major contributors to the pathogenesis of DM [2]. Specially, impaired pancreatic β-cell function is a prerequisite for the development of hyperglycemia and DM and progressive loss of β-cell function is associated with worsening hyperglycemia [3,4]. Insulin resistance, initially proposed to describe patients with diabetes requiring high doses of insulin [5], is currently defined as decreased responsiveness (maximal effect of insulin) or sensitivity (insulin concentration required for a half-maximal response) to metabolic actions of insulin, such as insulin- mediated glucose disposal of muscle and adipose tissue and inhibition of gluconeogenesis in liver [6]. Insulin resistance is also associated with metabolic syndrome, which includes dyslipidemia, hypertension, and obesity, resulting in an increased risk of cardiovascular disease [7]. Assessment of the pancreatic β-cell function and insulin resistance are very important to determine the type of DM and to plan an optimal management and preventive strategy in clinical practice. It is also of value in research fields such as new antidiabetic drug development, many kinds of experimental and clinical study on DM and metabolic disease. A variety of methods and indices have been developed to assess the β-cell function and insulin resistance/sensitivity based on static, dynamic test and calculation of their results. However, those remain challenging because of the complexity of the β-cell response to various stimuli, feedback mechanism and insulin signaling and metabolism in organs [8]. In order to solve the theoretical and practical difficulties, research on new index is still in progress. It is also important to understand the theoretical background, advantages, and limitations of each method to choose the appropriate method and index for the purpose of use and clinical situation.
- In this review, we summarized a variety of currently applicable methods and indices to evaluate pancreatic β-cell function and insulin resistance/sensitivity and introduced novel indices.
INTRODUCTION
- Insulin is secreted by pancreatic β-cell in response to plasma level of glucose and nutrients (amino acid, free fatty acid) and an additional signal like metabolic factor, neurotransmitter and hormone modulate insulin secretion [9]. Glucose is the major factor controlling β-cell function and survival. Glucose entering β-cell via glucose transporters is rapidly phosphorylated to glucose-6-phosphate by glucokinase and undergoes oxidation in mitochondria, leading to production of adenosine triphosphate (ATP). The rise of ATP/adenosine diphosphate ratio in β-cell leads to subsequent closure of the KATP channel which elicits cell membrane depolarization and allows the entry of Ca2+ through the opening of L-type voltage-dependent calcium channels. Raised levels of intracellular Ca2+ induce exocytosis of secretory granules containing insulin/proinsulin from pancreatic β-cell. The pharmacologic half‐life of insulin is estimated to be between 5 and 8 minutes, and is mainly cleared by insulinase activity within the liver, kidneys, and some other tissues [10].
- Glucose induces insulin secretion (GSIS) in a biphasic manner; a rapid, early peak (1st phase) is followed by a second slower and gradually rising peak (2nd phase) [11]. First phase insulin secretion is rapidly stimulated by the increased cytosolic Ca2+ and is largely due to the exocytosis of primed (readily releasable) insulin granules. Second phase insulin secretion is slow, activated by cytosolic Ca2+, ATP, and cyclic adenosine monophosphate production, and due to the subsequent supply of new insulin granules for release. GSIS can be modulated by amino acids, free fatty-acids and non-nutrient secretagogues, such as incretin hormones, growth factors, and neurotransmitters.
- Insulin binds to the plasma membrane-bound receptor of the target cell and coordinates the integrative anabolic action of nutrient availability [12]. The insulin receptor (IR), a tyrosine kinase, consists of α and β chains and is also activated by insulin-like growth factor I and II as well as insulin. There are two IR isoforms, A and B, but the B isoform is the primary isoform and more specific for insulin. The B isoform is expressed in liver, muscle, and adipose tissue, thus thought to mediate most metabolic effects of insulin. Binding of these endogenous ligands to the α chain of IR causes structural changes in the β chain by inducing auto‐phosphorylation in tyrosine residues. These changes are important for downstream events such as recruitment of the adaptor proteins IR substrates. The downstream of IR activation can be functionally divided into mitogenic and metabolic signals.
- The role of insulin in glucose homeostasis is represented by the direct effect of insulin on skeletal muscle, liver and white adipocytes [12]. These tissues require tissue-specific insulin signaling pathways. In skeletal muscle, a representative insulin-acting tissue, insulin promotes glucose utilization and storage by increasing glucose transport and net glycogen synthesis. In the liver, insulin activates glycogen synthesis, increases adipogenic gene expression, and inhibits gluconeogenesis by decreasing gluconeogenic gene expression. In white adipocyte tissue, insulin inhibits lipolysis and increases glucose transport and adipogenesis. Although the effects of insulin vary from tissue to tissue, the proximal components involved in insulin signaling are very similar in all insulin-responsive cells. Moreover, insulin suppresses glucagon secretion from pancreatic α-cells which is itself a potent inducer of hyperglycemia [13].
- The 1st phase insulin secretion of GSIS is known to be pivotal in the transition from fasting to feeding through the following functions: inhibition of hepatic glucose production (HGP); inhibition of lipolysis; and preparation of target cells for action of insulin across the endothelial barrier [14]. The 2nd phase insulin secretion of GSIS reduces HGP as in 1st phase, but to a lesser extent [15]. More important, it increases glucose utilization in peripheral tissues [16]. Therefore, although the importance of 2nd phase insulin secretion is relatively underestimated compared to that in the 1st phase, it is important for maintaining glucose homeostasis.
INSULIN SECRETION AND INSULIN ACTION
- To fully assess pancreatic β-cell function, both β-cell mass and insulin secretory function should be measured; however, β-cell mass cannot be directly measured in human. In general, β-cell function can be estimated by measuring insulin or C-peptide secreted with insulin from β-cells on an equimolar basis. About half of newly secreted insulin molecules are extracted in the first pass by the liver, and the extent of hepatic insulin clearance depends on the disease state and insulin load [17], in contrast, the C-peptide is not significantly cleared by the liver [18,19]. In vivo evaluation of dynamic β-cell insulin release from the hepatic portal vein [20] is rarely practical in humans, so this problem can be addressed by exploiting the properties of insulin and C-peptide that are co-secreted but have different clearance mechanisms [21].
- By mathematically modeling (with deconvolution) the time-sequentially measured C-peptide and insulin concentrations under various conditions that stimulate β-cells, the pre-hepatic insulin secretion rate (ISR) can be deduced [22].
- By the way, basal β-cell function is measured insulin or C-peptide in static situation but 1st phase, 2nd phase insulin secretion, the dose-response curve between glucose and ISR and maximal insulin secretory function can be measured in dynamic stimulatory condition. For this dynamic test, a single glucose injection (such as single bolus, fixed dose infusion or oral glucose intake), graded glucose infusion (GGI) or hyperglycemic clamp technique were introduced. Furthermore, non-glucose stimulator as glucagon or arginine is used to induce maximal insulin response during dynamic test.
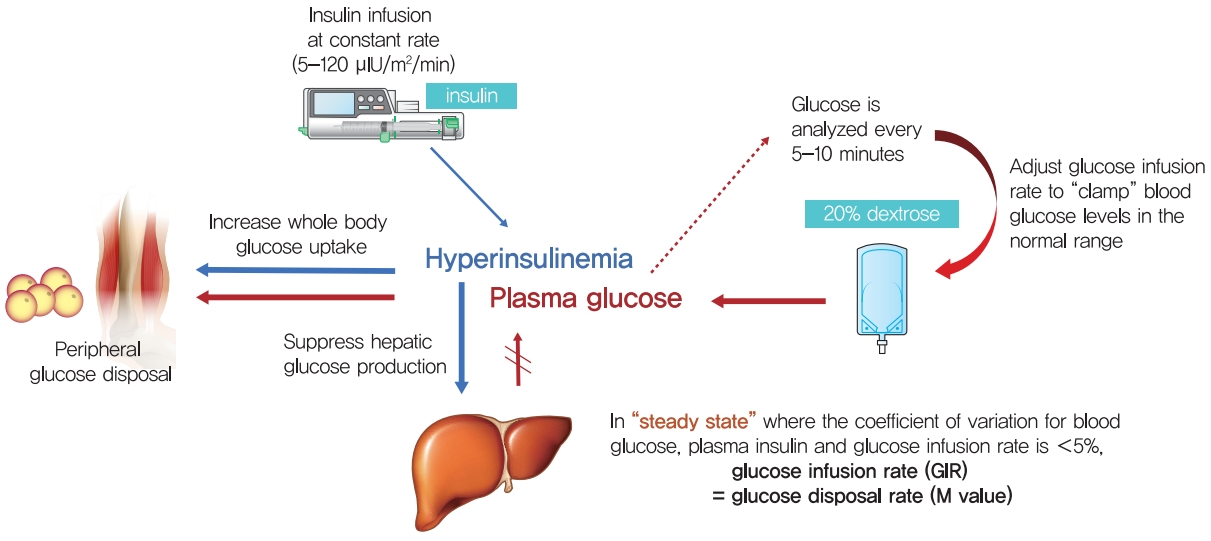
- In order to measure whole-body insulin action, specific conditions are required to completely suppress endogenous glucose production and to measure the glucose disposal of each tissue by insulin injected from the outside. Conditions to reach a new euglycemic steady state in the hyperinsulinemic state by infusion of various concentrations of insulin to suppress HGP (which is the main), and continuous glucose infusion to maintain the euglycemic status [23]. At this time, since glucose infusion rate (GIR) reflects the rate of glucose utilization in the body, it is possible to directly measure the systemic action of insulin on glucose utilization. Under these conditions, 80% to 90% of the infused glucose is consumed by skeletal muscle and less than 5% in adipose tissue and negligible in other tissues, so the measured insulin action mostly reflects the insulin sensitivity of skeletal muscle. Therefore, insulin sensitivity can be measured directly by using the hyperinsulinemic euglycemic clamp (HEC) method using these conditions.
- Because HEC studies are not feasible in usual clinical condition and studies, measurement of insulin sensitivity by the frequently sampled intravenous glucose tolerance test (FSIVGTT) can be used to assess both hepatic and peripheral insulin resistance and correlates well with the HEC technique [24]. Other surrogate markers of insulin sensitivity are calculated by glucose and insulin values in the fasting state or after an oral glucose tolerance test (OGTT) [25-27]. The homeostatic model assessment (HOMA) from insulin and glucose in the fasting state correlates well with the HEC study [27], but it primarily reflects hepatic insulin sensitivity, since the fasting plasma glucose is determined mainly by the rate of HGP and insulin is the primary regulator of HGP. Therefore, the correlation between HOMA and the HEC is less robust when analyzed in subjects with impaired glucose tolerance [28]. On the other hand, indexes of insulin resistance from the OGTT reflect both hepatic and peripheral insulin resistance [26]. Because significant (30% to 40%) amounts of glucose are taken up by the splanchnic bed during an OGTT and also HGP is less completely suppressed than during the HEC study [29], the plasma glucose concentration during OGTT is affected by both hepatic and peripheral (primarily muscle) insulin resistance. This OGTT-derived indexes correlate well (R; approximately 0.7) with insulin sensitivity measured with the HEC study [26].
- Intravenous glucose tolerance test
- After an overnight fast, an intravenous bolus of glucose (0.3 g/kg body weight) is infused over 2 minutes starting at time 0. Twenty minutes after glucose infusion, a certain amount of insulin is injected over 5 minutes. In the modified FSIVGTT, exogenous insulin (4 μIU/kg/min) is used [30-32]. Tolbutamide was used instead of insulin in the modified FSIVGTT to stimulate endogenous insulin secretion [32-35]. Blood samples are taken for plasma glucose and insulin measurements at −10, −1, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 16, 20, 22, 23, 24, 25, 27, 30, 40, 50, 60, 70, 80, 90, 100, 120, 160, and 180 minutes. Two phases of insulin release are observed during FSIVGTT. FSIVGTT can represent β-cell response to rising glucose level in the condition similar to eating. In FSIVGTT, glucose is intravenously injected and stimulate directly the β-cells. Because it does not have to pass through the digestive system, it is free from incretin effect and various gastric emptying time compared with OGTT. FSIVGTT is influenced by insulin and glucose clearance.
- Minimal model analysis was developed by Bergman et al. [36]. Data obtained from FSIVGTT are calculated using the computer program MINMOD to generate an index of β-cell function, insulin sensitivity and glucose effectiveness. C-peptide can be used instead of insulin in minimal model analysis with FSIVGTT.
- Minimal model analysis is defined by two couple differential equations, describing glucose dynamics in a mono-compartmental “glucose space” and insulin dynamics in a “remote compartment.” Minimal model uniquely identifies model parameters that determine a best fit to glucose disappearance during the modified FSIVGTT. Minimal model analysis is less labor-intensive than hyperglycemic clamp and does not require steady-state conditions, and does constant intravenous infusion rate control. But, minimal model analysis excessively simplifies the normal physiology of glucose metabolism, resulting in poor accuracy [37,38].
- Hyperglycemic clamp test
- In hyperglycemic clamp, a high target glucose concentration, which is enough to stimulate insulin secretion, would be maintained by infusion of glucose. A pre-determined priming dose of dextrose is infused for 15 minutes to raise the glucose concentration to a hyperglycemic target level in a short time, followed by an infusion of 20% dextrose for 120 minutes at variable rates to maintain plasma glucose levels at a constant level. Blood samples for plasma glucose and insulin are collected at baseline, every 2 minutes during the first 15 minutes, and then, every 5 to 10 minutes for glucose and insulin measurement during the test. Acute insulin response (AIR) during the first 10 minutes, late insulin response between 10 and 120 minutes are measured [39]. In the presence of hyperglycemia, glucagon (1 mg) can be administered intravenously at 120 minutes to induce an insulin response to non-glucose stimulation. After glucagon injection, additional blood samples are collected at 1, 3, 6, and 10 minutes [40]. The Arginine stimulation test can be performed to evaluate maximal β-cell function at the end of hyperglycemic clamp test [41]. After achieving a plasma glucose concentration up to 350 mg/dL by glucose infusion, baseline samples were taken at ±5 and ±2 minutes. A maximally stimulating dose of arginine hydrochloride (5 g) was then injected intravenously for 45 seconds. Samples were taken at 2, 3, 4, and 5 minutes after injection. Hyperglycemic clamp is a highly reproducible and reliable assessment of β-cell responsivity to glucose. However, this method has technical difficulty, and it is expensive and is time- and labor-intensive.
- Hyperinsulinemic euglycemic clamp test
- The HEC test, developed by DeFronzo et al. [39], is widely accepted as the gold standard for the direct determination of insulin resistance/sensitivity in humans [42]. Insulin is administered intravenously at a constant rate (5 to 120 μIU/m2/min) after an overnight fast. The new equilibrium insulin concentration is maintained at a higher concentration than in the fasting state. At this time, HGP is suppressed and glucose uptake in muscle and adipose tissue is increasing. In this state, blood glucose levels are frequently monitored at intervals of 5 to 10 minutes with a glucose analyzer, while 20% dextrose is administered intravenously to maintain a blood glucose level in the normal range (euglycemic). An infusion of potassium phosphate is also given to prevent hypokalemia resulting from hyperinsulinemia and increased glucose disposal. After several hours of insulin infusion at a constant rate, an equilibrium state is reached in which plasma insulin, blood glucose, and GIR are kept constant. “Steady-state” is defined as a period greater than 30-minute (at least 1 hour after initiation of insulin infusion) during which the coefficient of variation for blood glucose, plasma insulin, and GIR is less than 5% [6,43]. Assuming that hyperinsulinemia sufficiently inhibited HGP, the GIR is consistent with the glucose disposal rate (M) since there is no difference between the infused and utilized glucose under this equilibrium condition (Fig. 1) [43]. M is corrected for body weight, especially fat-free mass. Thus, the HEC can measure whole body glucose disposal (mainly in skeletal muscle) directly at a given level of hyperinsulinemia under steady-state. When radiolabeled glucose tracers are used under clamp conditions, it is possible to simultaneously quantify HGP as well as whole body glucose disposal [44,45]. When insulin sensitivity is increased, the administered glucose amount is increased. In this case, the difference in blood glucose level between the arterial and venous blood is also increased. Therefore, if the GIR is determined based on the concentration of glucose measured in venous blood, insulin sensitivity may be over-measured. To solve this problem, the hand used for blood sampling may be cannulated in a retrograde fashion and warmed with a heating pad (opening arteriovenous anastomoses) to “arterialize” the venous blood [46]. Radiolabeled tracers of glycerol or amino acids may be used during the HEC to assess insulin sensitivity with respect to lipolysis or protein metabolism [47,48]. 31P magnetic resonance spectroscopy may be used in conjunction with the HEC to assess rates of insulin-stimulated muscle mitochondrial ATP synthase flux and insulin-stimulated increases in concentrations of intramyocellular inorganic phosphate [49]. Doppler and contrast ultrasound imaging have also been used in conjunction with the HEC to study insulin sensitivity concerning vascular actions of insulin [50]. However, HEC study takes a lot of time and is a labor intensive and expensive. In addition, it is not suitable for use in large-scale epidemiologic studies and routine patient care because it requires a skilled operator. The HEC may not accurately reflect insulin action and glucose dynamics under physiological conditions that a dynamic test such as an oral meal or oral glucose load may determine.
- C-peptide minimal models with graded glucose infusion
- After an overnight fast, catheters are placed into both forearm for blood sample collection and glucose infusion. Glucose is administrated at progressively increasing and then decreasing rates (0, 4, 8, 16, 8, 4, 0 mg/kg/min). Each glucose infusion is administered for 40 minutes. Glucose and c-peptide levels are measured at 10-minute intervals during a 40-minute baseline period before the glucose infusion and throughout the 240-minute glucose infusion. Both basal and dynamic insulin secretory responses and β-cell sensitivity can be calculated using the C-peptide minimal models [51]. The ISR can be assessed using minimal model analysis computer software [52]. Because it is simpler than minimal model analysis with FSIVGTT, it can widely applied to clinical studies but the incretin effect is not reflected.
- Oral glucose tolerance test
- OGTT is the most widely used test in clinical practice to diagnose glucose intolerance and DM [53]. After overnight fast, blood samples for determinations of glucose and insulin concentrations are taken at 0, 30, 60, 90, and 120 minutes following a standard oral glucose load (75 g). β-cell function estimated from OGTT presents a more physiological pattern of the changes in glucose, insulin, and incretin hormones than the hyperglycemic clamp or FSIVGTT [21,54]. OGTT is easy to perform but cannot clearly reflect and distinguish the 1st and 2nd phases insulin secretion due to the variability in the glucose concentration.
- Mixed meal tolerance test
- Mixed meal tests were developed to reflect insulin and glucose responses to the various nutrients along with glucose. There are many kinds of mixed meal with various nutrient distribution composition (for example, 10 kcal/kg, 45% carbohydrate, 15% protein, and 40% fat). Blood samples were collected for glucose, insulin, and C-peptide concentration in fasting state and at 15, 30, 60, 90, 120, 180, and 240 minutes after mixed meal ingestion [53,55]. Like OGTT, the mixed meal tolerance test (MTT) mimics the glucose and insulin dynamics of physiological conditions more closely than the conditions of the hyperglycemic clamp, insulin suppression test (IST), or FSIVGTT. But, because of variation of glucose absorption and splanchnic glucose uptake, mixed MTT has poor reproducibility.
- Insulin suppression test
- The IST is used to directly measure insulin resistance. It was developed by Shen et al. [56] and modified by Harano et al. [57]. After an overnight fast, somatostatin (250 μg/hr) or the somatostatin analog octreotide (25 μg bolus, followed by 0.5 μg/min) is intravenously infused to suppress endogenous secretion of insulin and glucagon. It is assumed that the somatostatin infusion is sufficient to completely suppress endogenous insulin and glucagon secretion. A constant dose of insulin (25 μIU/m2 ∙min) and glucose (240 mg/m2 ∙min) is then infused into the same antecubital vein for 3 hours. From the contralateral arm, blood samples for glucose and insulin are collected every 30 minutes for 2.5 hours and then at 10-minute intervals from 150 to 180 minutes of the IST. Steady-state conditions are more easily achieved with the IST than with the HEC because of the lack of variable infusions. In individuals with marked insulin sensitivity, hypoglycemia may occur during the IST, and in individuals with type 2 DM, glucosuria may occur due to hyperglycemia, making it difficult to accurately measure insulin resistance. Somatostatin infusion can independently affect peripheral glucose clearance [58]. Therefore, IST is limited application in a large-scale epidemiologic studies or clinical trials.
- Glucagon stimulation test
- Glucagon stimulation test (GST) was described in 1977 [59]. Baseline serum C-peptide and plasma glucose sample are collected. An 1 mg glucagon is injected over 10 seconds. Samples are collected at 2, 4, and 6 minutes after glucagon administration. GST is a more sensitive and reproducible test compared to provocation test using other substrates, such as glucose or tolbutamide. And it is associated with diabetes type and future use of insulin. The majority of patients experienced nausea with the GST.
METHODS TO ASSESS PANCREATIC β-CELL FUNCTION/INSULIN RESISTANCE
- First- and second-phase insulin secretion
- During FSIVGTT, the 1st phase insulin secretion or AIR is represented as insulin release during the first 10 minutes after glucose injection [60], and calculated using the area under the curve (AUC) of insulin concentration during the first 10 minutes or mean of the 3, 4, and 5 minutes insulin values after glucose injection minus the basal value. Insulin secretion peaks in 3 to 10 minutes, at which time 40 μIU/mL or less is a low response, and more than 140 μIU/mL is a high response. The 2nd phase insulin secretion is calculated as the insulin area above the pre-stimulus level during 10 to 60 minutes after glucose bolus [61].
- In minimal model analysis with FSIVGTT, first phase β-cell responsivity (Φ1) is calculated amount of insulin released during the first peak in proportion to change in the glucose level. second phase β-cell responsivity (Φ2) is calculated as amount of insulin released during the second phase.
- During hyperglycemic clamp, AIR is AUC of insulin concentration during the first 10 minutes. And late insulin response is AUC of insulin concentration between 10 and 120 minutes. The acute insulin response to arginine (AIRarg) was calculated as the mean of the 2, 3, 4, and 5 minutes values minus the mean of the baseline values [41]. The AIRarg during the 230 mg/dL of glucose clamp allows for determination of the glucose-potentiation of arginine-induced insulin release (AIRpot). The AIRarg during the 340 mg/dL of glucose clamp allows for determination of the maximal potentiation of arginine-induced insulin release (AIRmax) since the acute responses to arginine are maximal at plasma glucose concentrations 315 mg/dL.
- C-peptide
- Because the half-life of C-peptide is longer (20 to 30 minutes) than that of insulin (3 to 5 minutes), and hepatic clearance of C-peptide is negligible, and clearance rate in the peripheral circulation is constant compared with insulin [62]. In healthy subjects, the plasma concentration of C-peptide in the fasting state is 0.3 to 0.6 nmol/L, with a postprandial increase to 1 to 3 nmol/L [63]. Spot or 24 hours urinary C-peptide, urinary C-peptide to creatinine ratio can be performed. C-peptide represents endogenous insulin production, and measurement of Cpeptide does not have the pitfall of cross-reaction of assay between exogenous and endogenous insulin in insulin-treated patients with diabetes. C-peptide also correlates with type of disease, duration of diabetes, as well as age of diagnosis. Because the majority of C-peptide is metabolized by the kidneys and then excreted in urine [64], C-peptide measurement is inaccurate in patients with chronic kidney disease. And C-peptide metabolism is influenced by age, sex, obesity, body shape, and degree of glucose tolerance [65]. The detection of subtle levels of C-peptide limited.
- In addition, fasting or postprandial C-peptide to glucose ratio (CPRI) was suggested as a practical marker of β-cell function in diabetes, especially, postprandial CPRI in type 2 DM [66]. C-peptide concentrations were markedly different and higher in the up and down GGI compared with FSIVGTT, it represents an effect of the glucose perturbation or statin and dynamic glucose control. β-Cells are more sensitive to a slow glucose increase, but β-cells response to a decreasing glucose stimulus.
- Insulinogenic index
- The insulinogenic index (IGI) is used to quantify the β-cell response according to the change of plasma glucose level. IGI is calculated from change in insulin/change in glucose over the first 30 minutes after the load on OGTT.
- IGI= [Δinsulin (30–0 minutes) (μIU/mL)/Δglucose (30–0 minutes) (mg/dL)]
- IGI is also calculated from a mixed MTT. β-Cell function parameters were higher during a mixed MTT than during an OGTT, it suggests that β-cell function parameters are influenced by meal composition [67].
- Homeostatic model assessment of β-cell function
- HOMA was developed in 1985 as a model for the correlation between glucose and insulin dynamics [27]. Both the original HOMA and the updated HOMA2 assume a feedback loop between the liver and β-cell. For example, glucose concentration is regulated by HGP via insulin, and insulin concentration is regulated by the response of pancreatic β-cells to glucose concentration. It can be used as an index representing insulin resistance and β-cell function. Homeostatic model assessment of β-cell function (HOMA-β) is a simple static assessment of β-cell function using basal values of glucose and insulin [27].
- HOMA-β= [fasting plasma insulin (μIU/mL)×360/(fasting plasma glucose (mg/dL)–63]
- Because HOMA-β is easy and simple, it is widely used in epidemiologic studies. HOMA-β has a limitation because fasting level of glucose and insulin at a single time point cannot represent the complex relationship between variables in glucose-insulin feedback system. And accurate insulin assay is required for reliable HOMA-β value [42].
- Disposition index
- Disposition index (DI) is expressed by calculating the product of insulin secretory capacity and insulin sensitivity. Alternatively, it is calculated by dividing the insulin secretory capacity by the insulin resistance value [68]. DI is the concept of insulin secretion capacity taking into account insulin resistance. In healthy subjects, the product of insulin sensitivity and insulin secretion is constant. Insulin secretion increases to response to increasing the insulin resistance, but when β-cell failure occurs, the capacity to compensate for insulin resistance decreases, resulting in lower DI. Thus, DI does not simply indicate insulin secretion but is considered to be a more useful and practical indicator of β-cell function by indicating the insulin secretory ability reflecting the insulin resistance state of each subject.
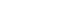
- The index for measuring β-cell function described so far is summarized in Table 1. According to the measurement method, it is classified into a dynamic test and a static test, and the calculation formulas for various insulin secretion indices calculated based on the values of insulin, C-peptide, and glucose measured through each test are summarized. In order to evaluate acute and late insulin secretory function and maximum insulin secretory capacity, dynamic test-based measurement is required. An appropriate method should be selected and utilized according to the strengths and weaknesses of the indices, the environment where they can be used, and the purpose.
INDICES OF PANCREATIC β-CELL FUNCTION
- Insulin sensitivity index
- Insulin sensitivity index (SI) is calculated form HEC.
- SIClamp=M/(G×ΔI) [25]
- M: glucose disposal rate,
- G: steady state blood glucose concentration,
- ΔI: difference between fasting and steady-state plasma insulin (SSPI) concentrations
- For accurate measurement for M and SIClamp in HEC test, it is important that the assumption that HGP is completely suppressed by steady-state hyperinsulinemia is met [25,45,69,70].
- Steady-state plasma glucose concentrations
- During IST, the period from 150 to 180 minutes is regarded as the steady-state period, and at this time, the constant infusions of insulin and glucose will determine the SSPI and steady-state plasma glucose (SSPG) concentrations. SSPI concentrations are generally similar among subjects, while the SSPG concentrations are inversely related to insulin sensitivity. Thus, SSPG concentration will be higher in insulin-resistant subjects and lower in insulin-sensitive subjects, which can be used as an indicator of insulin resistance/sensitivity. The SSPG is a highly reproducible direct measurement of metabolic actions of insulin, which is less labor-intensive and less technically demanding than the HEC.
- Insulin sensitivity (SI) and glucose effectiveness (SG)
- Insulin sensitivity (SI) and glucose effectiveness (SG) is calculated from minimal model analysis. SI is defined as fractional glucose disappearance per insulin concentration unit. SG is defined as the ability of glucose per se to promote its own disposal and inhibit HGP in the absence of an incremental insulin effect [68,71,72].
- 1/Fasting insulin
- 1/Fasting insulin is well known as a surrogate indicator of insulin resistance, which decreases as an individual’s insulin resistance increases [73]. However, because insulin concentrations are not normally distributed, linear correlation between 1/(fasting insulin) and insulin resistance from HEC method are not strong. And insulin secretion is decreased in hyperglycemic condition in diabetic subjects or glucose-intolerant subjects, which may cause erroneous results. Thus, 1/(fasting insulin) had limited use in patients with glucose intolerance or diabetes. There is a lack of standardized insulin assays.
- Glucose/insulin ratio
- The fasting glucose/insulin ratio (G/I ratio) was used as an index of insulin resistance/sensitivity in previous studies [74-76]. However, the G/I ratio does not appropriately reflect the physiology underlying the determinants of insulin sensitivity [77]. In healthy subjects, fasting G/I ratio is functionally equivalent to 1/(fasting glucose) due to fasting glucose levels in the normal range. But, in condition of relative fasting hyperinsulinemia in patients with glucose intolerance or diabetes, fating G/I ratio erroneously increases compared to 1/(fasting glycose). In patients with diabetes, the fasting insulin concentration is low and the maintaining blood glucose in the normal range is insufficient; thus, there are limitations in applying these surrogate indices derived to patients with diabetes.
- Homeostatic model assessment of insulin resistance
- HOMA-IR= [fasting insulin (μIU/mL)]×[fasting glucose (mmol/L)]/22.5
- Homeostasis model of insulin resistance (HOMA-IR) is simple, minimally invasive, but insulin resistance in patients treated with insulin needs further validation. The denominator of 22.5 is a normalizing factor. HOMA-IR has a reasonable linear correlation with HEC and minimal model estimates of insulin resistance in several studies of distinct populations [78, 79]. Log (HOMA-IR) represent a much stronger linear correlation with HEC estimates of insulin resistance. HOMA-IR or log (HOMA-IR) may not give appropriate results in subjects with severely impaired or absent β-cell function.
- Quantitative insulin sensitivity check index
- The quantitative insulin sensitivity check index (QUICKI) is an empirically derived mathematical transformation of fasting blood glucose and plasma insulin concentrations that provides a reliable, reproducible, and accurate index of insulin sensitivity with excellent positive predictive power [25,70,80,81]. It is simply a variation of the HOMA equation as it transforms the data by taking both the logarithm and the reciprocal of the glucose-insulin product. Since fasting insulin levels do not have a normal distribution, log transformation improves its linear correlation with SIClamp. In order to solve the problem that it is difficult to maintain euglycemia in diabetic patients with fasting hyperglycemia and impaired β-cell function, addition of log (fasting glucose) to log (fasting insulin) provides a reasonable correction.
- QUICKI= 1/[log (fasting insulin, μIU/mL)+log (fasting glucose, mg/dL)]
- QUICKI has a substantially better linear correlation with SIclamp than SI derived from the minimal model or HOMA-IR, especially in obese and diabetic subjects [25,70,81]. In addition, QUICKI has the strongest positive predictive power for determining development of diabetes [82], and is most correlated with changes in SIClamp after therapeutic interventions [70]. QUICKI is a simple, inexpensive, and non-invasive surrogate index. Compared with other surrogate indexes of insulin resistance, QUICKI is the most thoroughly evaluated and validated.
- McAuley index
- McAuley index [83] is used for predicting insulin resistance in normoglycemic individuals. Statistically, regression analysis method was used to estimate the cut-off values and the importance of various parameter for insulin resistance such as fasting insulin, triglycerides, aspartate aminotransferase, basal metabolic rate (body mass index [BMI]), and waist circumference.
- McAuley index=exp [2.63–0.28 ln (insulin in μIU/L)–0.31 ln (triglycerides in mmol/L)]
- Matsuda index
- The composite whole-body insulin SI including both hepatic and peripheral tissue insulin sensitivity developed by Matsuda and DeFronzo from OGTT data [26].
- ISI(Matsuda)=10,000/√[(G0×I0) ×(Gmean×Imean)]
- G0: Fasting plasma glucose concentration (mg/dL) taken from time 0 of the OGTT
- I0: Fasting plasma insulin concentration (μIU/L) taken from time 0 of the OGTT
- Gmean: Mean plasma glucose concentration during OGTT (mg/dL)
- Imean: Mean plasma insulin concentration during OGTT (μIU/L)
- The square root is used to correct for nonlinear distribution of insulin, and 10,000 is a scaling factor in the equation.
- Stumvoll index
- Stumvoll and Gerich proposed use of demographic data such as age, sex, and BMI in addition to plasma glucose (mmol/L) and insulin (pmol/L) responses during the OGTT to predict insulin sensitivity and β-cell function [84].
- ISIStumvoll=0.156–0.0000459×I120(pmol/L)–0.000321×I0(pmol/L)–0.00541×G120(mmol/L)
- I0, plasma insulin concentration during fasting
- I120, plasma insulin concentration at 120 minutes
- G120, plasma glucose concentration at 120 minutes
- Avignon index
- Avignon et al. [85] developed three insulin sensitivity indicies: SiM, Sib, and Si2h by computation of fasting and 2-hour glucose and insulin data during OGTT. It is useful for blending both the determination of glucose tolerance and an estimate of insulin sensitivity in a single and simple test.
- Sib=108/(I0(μIU/L)×G0(mmol/l)×VD)
- Si2h=108/(I120(μIU/l)×G120(mmol/l)×VD)
- SiM=[(0.137×Sib)+Si2h]/2
- I0, plasma insulin concentration during fasting
- I120, plasma insulin concentration at 120 minutes
- G0, plasma glucose concentration during fasting
- G120, plasma glucose concentration at 120 minutes
- VD, glucose distribution volume (150 mL/kg body weight)
- Gutt index, ISI (0,120)
- ISI (0,120) was calculated using the plasma glucose and insulin concentration from fasting (0 minute) and 120-minute samples from the OGTT [86]. The index correlates well with direct estimates of insulin sensitivity obtained from the HEC study.
- ISI(0, 120)=75,000+(G0–G120)(mmol/L)×0.19×body weight(kg)/120×Gmean(0, 120)(mmol/L)×Log (Imean(0, 120))(μIU/L)
- G0, plasma glucose concentration during fasting during OGTT
- G120, plasma glucose concentration at 120 minutes during OGTT
- Gmean, mean plasma glucose concentration during OGTT
- Imean, mean insulin concentration during OGTT
- Belfiore index
- The Belfiore index was calculated with the normal value for basal glucose and insulin concentrations, and for mean normal value for glucose and insulin areas during OGTT [87].
- Belfiore index=2(GS/GN)×(IS/IN)+1
- S, subjects
- N, normal reference values
- GS, GN: plasma glucose concentrations expressed as fasting values or as areas obtained during a standard OGTT at 0 and 2 hours
- IS, IN: plasma insulin concentrations expressed as fasting values or as areas obtained during a standard OGTT at 0 and 2 hours
- The indices for measuring insulin sensitivity/resistance described so far are summarized in Table 2. Direct measures of insulin sensitivity based on HEC or IST mainly reflect insulin sensitivity of skeletal muscle. On the other hand, indirect measures of insulin sensitivity based on FSIVGTT or surrogate indices based on OGTT or MTT reflect hepatic and peripheral insulin resistance. Static test-based insulin resistance indices primarily reflect hepatic insulin sensitivity.
INDICES OF INSULIN INSENSITIVITY/RESISTANCE
- To date, various methods have been developed to measure pancreatic β-cell function and insulin resistance/sensitivity. Each of these tests has advantages and limitations. The most appropriate method should be selected in consideration of the purposes, methods, and the practical situation. The hyperglycemic clamp and HEC study are still the gold standard methods for β-cell function and insulin sensitivity respectively, but these are invasive and labor intensive. Thus, it is necessary to develop a non-invasive, inexpensive, and easy method that best reflects the physiology of glucose and insulin metabolism. A better understanding about the how to measure β-cell function and insulin resistance/sensitivity will help us understand the pathophysiology of DM and further develop the treatment strategy and new drugs.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
This research was funded by the Korea Disease Control and Prevention Agency, grant number 2020-ER6402-00.
NOTES
-
Acknowledgements
- None

FSIVGTT, frequently sampled intravenous glucose tolerance test; AIR, acute insulin response; AUC, area under the curve; DI, disposition index; SRS, secretion rate by glucose concentration (static glucose control); SRd, secretion rate by the rate of change of glucose concentration (dynamic glucose control); OGTT, oral glucose tolerance test; MTT, meal tolerance test; IGI, insulinogenic index; HOMA-IR, homeostatic model assessment of insulin resistance; HOMA-β, homeostatic model assessment of β-cell function.
SI, insulin sensitivity index; M, glucose disposal rate; G, steady state blood glucose concentration; ΔI, difference between fasting and steady-state plasma insulin concentrations; FSIVGTT, frequently sampled intravenous glucose tolerance test; HGP, hepatic glucose production; OGTT, oral glucose tolerance test; MTT, meal tolerance test; ISI, insulin sensitivity index; Gmean, mean plasma glucose concentration during OGTT; Imean, mean insulin concentration during OGTT; G0, plasma glucose concentration during fasting; G120, plasma glucose concentration at 120 minutes; I0, plasma insulin concentration during fasting (μIU/L); I120, plasma insulin concentration at 120 minutes; HOMA-IR, homeostatic model assessment of insulin resistance; QUICKI, quantitative insulin sensitivity check index; TAG0, fasting triglyceride concentration.
- 1. Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus: present and future perspectives. Nat Rev Endocrinol 2011;8:228-36.ArticlePubMedPDF
- 2. DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 2004;88:787-835.ArticlePubMed
- 3. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837-53.ArticlePubMed
- 4. Viberti G, Kahn SE, Greene DA, Herman WH, Zinman B, Holman RR, et al. A diabetes outcome progression trial (ADOPT): an international multicenter study of the comparative efficacy of rosiglitazone, glyburide, and metformin in recently diagnosed type 2 diabetes. Diabetes Care 2002;25:1737-43.PubMed
- 5. Moller DE, Flier JS. Insulin resistance: mechanisms, syndromes, and implications. N Engl J Med 1991;325:938-48.ArticlePubMed
- 6. Muniyappa R, Lee S, Chen H, Quon MJ. Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage. Am J Physiol Endocrinol Metab 2008;294:E15-26.ArticlePubMed
- 7. Reaven G. The metabolic syndrome or the insulin resistance syndrome?: different names, different concepts, and different goals. Endocrinol Metab Clin North Am 2004;33:283-303.ArticlePubMed
- 8. Nielsen JH, Galsgaard ED, Moldrup A, Friedrichsen BN, Billestrup N, Hansen JA, et al. Regulation of beta-cell mass by hormones and growth factors. Diabetes 2001;50 Suppl 1:S25-9.ArticlePubMedPDF
- 9. Newsholme P, Krause M. Nutritional regulation of insulin secretion: implications for diabetes. Clin Biochem Rev 2012;33:35-47.PubMedPMC
- 10. Koeppen BM, Stanton BA. Berne & Levy physiology. 7th ed. Philadelphia: Elsevier; 2018.
- 11. Rorsman P, Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003;46:1029-45.ArticlePubMedPDF
- 12. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev 2018;98:2133-223.ArticlePubMedPMC
- 13. Faerch K, Vistisen D, Pacini G, Torekov SS, Johansen NB, Witte DR, et al. Insulin resistance is accompanied by increased fasting glucagon and delayed glucagon suppression in individuals with normal and impaired glucose regulation. Diabetes 2016;65:3473-81.ArticlePubMedPDF
- 14. Chon S, Gautier JF. An update on the effect of incretin-based therapies on β-cell function and mass. Diabetes Metab J 2016;40:99-114.ArticlePubMedPMCPDF
- 15. Luzi L, DeFronzo RA. Effect of loss of first-phase insulin secretion on hepatic glucose production and tissue glucose disposal in humans. Am J Physiol 1989;257(2 Pt 1):E241-6.ArticlePubMed
- 16. Steiner KE, Mouton SM, Bowles CR, Williams PE, Cherrington AD. The relative importance of first- and second-phase insulin secretion in countering the action of glucagon on glucose turnover in the conscious dog. Diabetes 1982;31:964-72.ArticlePubMedPDF
- 17. Hansen BC, Vinik A, Jen KL, Schielke GP. Fluctuations in basal levels and effects of altered nutrition on plasma somatostatin. Am J Physiol 1982;243:R289-95.ArticlePubMed
- 18. Ferrannini E, Cobelli C. The kinetics of insulin in man. II. Role of the liver. Diabetes Metab Rev 1987;3:365-97.ArticlePubMed
- 19. Ferrannini E, Cobelli C. The kinetics of insulin in man. I. General aspects. Diabetes Metab Rev 1987;3:335-63.ArticlePubMed
- 20. Hansen BC, Striffler JS, Bodkin NL. Decreased hepatic insulin extraction precedes overt noninsulin dependent (type II) diabetes in obese monkeys. Obes Res 1993;1:252-60.ArticlePubMed
- 21. Hovorka R, Jones RH. How to measure insulin secretion. Diabetes Metab Rev 1994;10:91-117.ArticlePubMed
- 22. Hovorka R, Soons PA, Young MA. ISEC: a program to calculate insulin secretion. Comput Methods Programs Biomed 1996;50:253-64.ArticlePubMed
- 23. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009;32(Suppl 2):S157-63.ArticlePubMedPMCPDF
- 24. Pacini G, Finegood DT, Bergman RN. A minimal-model-based glucose clamp yielding insulin sensitivity independent of glycemia. Diabetes 1982;31(5 Pt 1):432-41.ArticlePubMed
- 25. Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 2000;85:2402-10.ArticlePubMedPDF
- 26. Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 1999;22:1462-70.ArticlePubMedPDF
- 27. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412-9.ArticlePubMedPDF
- 28. Tripathy D, Almgren P, Tuomi T, Groop L. Contribution of insulin-stimulated glucose uptake and basal hepatic insulin sensitivity to surrogate measures of insulin sensitivity. Diabetes Care 2004;27:2204-10.ArticlePubMedPDF
- 29. Ferrannini E, Bjorkman O, Reichard GA Jr, Pilo A, Olsson M, Wahren J, et al. The disposal of an oral glucose load in healthy subjects: a quantitative study. Diabetes 1985;34:580-8.ArticlePubMed
- 30. Finegood DT, Hramiak IM, Dupre J. A modified protocol for estimation of insulin sensitivity with the minimal model of glucose kinetics in patients with insulin-dependent diabetes. J Clin Endocrinol Metab 1990;70:1538-49.ArticlePubMed
- 31. Quon MJ, Cochran C, Taylor SI, Eastman RC. Direct comparison of standard and insulin modified protocols for minimal model estimation of insulin sensitivity in normal subjects. Diabetes Res 1994;25:139-49.PubMed
- 32. Saad MF, Steil GM, Kades WW, Ayad MF, Elsewafy WA, Boyadjian R, et al. Differences between the tolbutamide-boosted and the insulin-modified minimal model protocols. Diabetes 1997;46:1167-71.ArticlePubMed
- 33. Beard JC, Bergman RN, Ward WK, Porte D Jr. The insulin sensitivity index in nondiabetic man. Correlation between clampderived and IVGTT-derived values. Diabetes 1986;35:362-9.ArticlePubMed
- 34. Bergman RN, Prager R, Volund A, Olefsky JM. Equivalence of the insulin sensitivity index in man derived by the minimal model method and the euglycemic glucose clamp. J Clin Invest 1987;79:790-800.ArticlePubMedPMC
- 35. Yang YJ, Youn JH, Bergman RN. Modified protocols improve insulin sensitivity estimation using the minimal model. Am J Physiol 1987;253(6 Pt 1):E595-602.ArticlePubMed
- 36. Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol 1979;236:E667-77.ArticlePubMed
- 37. Cobelli C, Bettini F, Caumo A, Quon MJ. Overestimation of minimal model glucose effectiveness in presence of insulin response is due to undermodeling. Am J Physiol 1998;275:E1031-6.ArticlePubMed
- 38. Cobelli C, Caumo A, Omenetto M. Minimal model SG overestimation and SI underestimation: improved accuracy by a Bayesian two-compartment model. Am J Physiol 1999;277:E481-8.ArticlePubMed
- 39. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214-23.ArticlePubMed
- 40. Choi CS, Kim C, Lee WJ, Park JY, Hong SK, Lee MG, et al. Association between birth weight and insulin sensitivity in healthy young men in Korea: role of visceral adiposity. Diabetes Res Clin Pract 2000;49:53-9.ArticlePubMed
- 41. Larsson H, Ahren B. Glucose-dependent arginine stimulation test for characterization of islet function: studies on reproducibility and priming effect of arginine. Diabetologia 1998;41:772-7.ArticlePubMedPDF
- 42. Choi CS, Kim MY, Han K, Lee MS. Assessment of β-cell function in human patients. Islets 2012;4:79-83.ArticlePubMed
- 43. Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, et al. Endotext. South Dartmouth: MDText.com Inc; 2021 Chapter, Assessing insulin sensitivity and resistance in humans [cited 2021 Sep 11]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK278954.
- 44. Campbell PJ, Mandarino LJ, Gerich JE. Quantification of the relative impairment in actions of insulin on hepatic glucose production and peripheral glucose uptake in non-insulin-dependent diabetes mellitus. Metabolism 1988;37:15-21.ArticlePubMed
- 45. Rizza RA, Mandarino LJ, Gerich JE. Dose-response characteristics for effects of insulin on production and utilization of glucose in man. Am J Physiol 1981;240:E630-9.ArticlePubMed
- 46. Morris AD, Ueda S, Petrie JR, Connell JM, Elliott HL, Donnelly R. The euglycaemic hyperinsulinaemic clamp: an evaluation of current methodology. Clin Exp Pharmacol Physiol 1997;24:513-8.ArticlePubMed
- 47. Gelfand RA, Barrett EJ. Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. J Clin Invest 1987;80:1-6.ArticlePubMedPMC
- 48. Greenfield MS, Doberne L, Kraemer F, Tobey T, Reaven G. Assessment of insulin resistance with the insulin suppression test and the euglycemic clamp. Diabetes 1981;30:387-92.ArticlePubMed
- 49. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 2004;350:664-71.ArticlePubMedPMC
- 50. Clerk LH, Vincent MA, Jahn LA, Liu Z, Lindner JR, Barrett EJ. Obesity blunts insulin-mediated microvascular recruitment in human forearm muscle. Diabetes 2006;55:1436-42.ArticlePubMedPDF
- 51. Toffolo G, Breda E, Cavaghan MK, Ehrmann DA, Polonsky KS, Cobelli C. Quantitative indexes of beta-cell function during graded up&down glucose infusion from C-peptide minimal models. Am J Physiol Endocrinol Metab 2001;280:E2-10.PubMed
- 52. Eaton RP, Allen RC, Schade DS, Erickson KM, Standefer J. Prehepatic insulin production in man: kinetic analysis using peripheral connecting peptide behavior. J Clin Endocrinol Metab 1980;51:520-8.ArticlePubMed
- 53. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2007;30 Suppl 1:S42-7.ArticlePubMedPDF
- 54. Yasuhara D, Naruo T, Nagai N, Tanaka M, Muranaga T, Nozoe S. Insulinogenic index at 15 min as a marker of nutritional rehabilitation in anorexia nervosa. Am J Clin Nutr 2003;77:292-9.ArticlePubMed
- 55. Dalla Man C, Campioni M, Polonsky KS, Basu R, Rizza RA, Toffolo G, et al. Two-hour seven-sample oral glucose tolerance test and meal protocol: minimal model assessment of beta-cell responsivity and insulin sensitivity in nondiabetic individuals. Diabetes 2005;54:3265-73.PubMed
- 56. Shen SW, Reaven GM, Farquhar JW. Comparison of impedance to insulin-mediated glucose uptake in normal subjects and in subjects with latent diabetes. J Clin Invest 1970;49:2151-60.ArticlePubMedPMC
- 57. Harano Y, Hidaka H, Takatsuki K, Ohgaku S, Haneda M, Motoi S, et al. Glucose, insulin, and somatostatin infusion for the determination of insulin sensitivity in vivo. Metabolism 1978;27(9 Suppl 1):1449-52.ArticlePubMed
- 58. Bergman RN, Ader M, Finegood DT, Pacini G. Extrapancreatic effect of somatostatin infusion to increase glucose clearance. Am J Physiol 1984;247(3 Pt 1):E370-9.ArticlePubMed
- 59. Faber OK, Binder C. C-peptide response to glucagon: a test for the residual beta-cell function in diabetes mellitus. Diabetes 1977;26:605-10.ArticlePubMed
- 60. Pratley RE, Weyer C. The role of impaired early insulin secretion in the pathogenesis of type II diabetes mellitus. Diabetologia 2001;44:929-45.ArticlePubMedPDF
- 61. Pfeifer MA, Graf RJ, Halter JB, Porte D Jr. The regulation of glucose-induced insulin secretion by pre-stimulus glucose level and tolbutamide in normal man. Diabetologia 1981;21:198-205.ArticlePubMedPDF
- 62. Leighton E, Sainsbury CA, Jones GC. A practical review of cpeptide testing in diabetes. Diabetes Ther 2017;8:475-87.ArticlePubMedPMCPDF
- 63. Yosten GL, Maric-Bilkan C, Luppi P, Wahren J. Physiological effects and therapeutic potential of proinsulin C-peptide. Am J Physiol Endocrinol Metab 2014;307:E955-68.ArticlePubMedPMC
- 64. Jones AG, Hattersley AT. The clinical utility of C-peptide measurement in the care of patients with diabetes. Diabet Med 2013;30:803-17.ArticlePubMedPMCPDF
- 65. Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels: comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992;41:368-77.ArticlePubMed
- 66. Saisho Y. Postprandial C-peptide to glucose ratio as a marker of β cell function: implication for the management of type 2 diabetes. Int J Mol Sci 2016;17:744.ArticlePMC
- 67. Rijkelijkhuizen JM, Girman CJ, Mari A, Alssema M, Rhodes T, Nijpels G, et al. Classical and model-based estimates of betacell function during a mixed meal vs. an OGTT in a population-based cohort. Diabetes Res Clin Pract 2009;83:280-8.ArticlePubMed
- 68. Bergman RN, Ader M, Huecking K, Van Citters G. Accurate assessment of beta-cell function: the hyperbolic correction. Diabetes 2002;51 Suppl 1:S212-20.PubMed
- 69. Bonadonna RC, Groop L, Kraemer N, Ferrannini E, Del Prato S, DeFronzo RA. Obesity and insulin resistance in humans: a dose-response study. Metabolism 1990;39:452-9.ArticlePubMed
- 70. Chen H, Sullivan G, Yue LQ, Katz A, Quon MJ. QUICKI is a useful index of insulin sensitivity in subjects with hypertension. Am J Physiol Endocrinol Metab 2003;284:E804-12.ArticlePubMed
- 71. Bergman RN. Orchestration of glucose homeostasis: from a small acorn to the California oak. Diabetes 2007;56:1489-501.PubMed
- 72. Haffner SM, Howard G, Mayer E, Bergman RN, Savage PJ, Rewers M, et al. Insulin sensitivity and acute insulin response in African-Americans, non-Hispanic whites, and Hispanics with NIDDM: the Insulin Resistance Atherosclerosis Study. Diabetes 1997;46:63-9.ArticlePubMed
- 73. Laakso M. How good a marker is insulin level for insulin resistance? Am J Epidemiol 1993;137:959-65.ArticlePubMed
- 74. Legro RS, Finegood D, Dunaif A. A fasting glucose to insulin ratio is a useful measure of insulin sensitivity in women with polycystic ovary syndrome. J Clin Endocrinol Metab 1998;83:2694-8.ArticlePubMed
- 75. Silfen ME, Manibo AM, McMahon DJ, Levine LS, Murphy AR, Oberfield SE. Comparison of simple measures of insulin sensitivity in young girls with premature adrenarche: the fasting glucose to insulin ratio may be a simple and useful measure. J Clin Endocrinol Metab 2001;86:2863-8.ArticlePubMed
- 76. Vuguin P, Saenger P, Dimartino-Nardi J. Fasting glucose insulin ratio: a useful measure of insulin resistance in girls with premature adrenarche. J Clin Endocrinol Metab 2001;86:4618-21.ArticlePubMed
- 77. Quon MJ. Limitations of the fasting glucose to insulin ratio as an index of insulin sensitivity. J Clin Endocrinol Metab 2001;86:4615-7.ArticlePubMed
- 78. Radziuk J. Insulin sensitivity and its measurement: structural commonalities among the methods. J Clin Endocrinol Metab 2000;85:4426-33.ArticlePubMed
- 79. Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care 2004;27:1487-95.ArticlePubMedPDF
- 80. Chen H, Sullivan G, Quon MJ. Assessing the predictive accuracy of QUICKI as a surrogate index for insulin sensitivity using a calibration model. Diabetes 2005;54:1914-25.ArticlePubMedPDF
- 81. Mather KJ, Hunt AE, Steinberg HO, Paradisi G, Hook G, Katz A, et al. Repeatability characteristics of simple indices of insulin resistance: implications for research applications. J Clin Endocrinol Metab 2001;86:5457-64.ArticlePubMed
- 82. Hanley AJ, Williams K, Gonzalez C, D’Agostino RB Jr, Wagenknecht LE, Stern MP, et al. Prediction of type 2 diabetes using simple measures of insulin resistance: combined results from the San Antonio Heart Study, the Mexico City Diabetes Study, and the Insulin Resistance Atherosclerosis Study. Diabetes 2003;52:463-9.PubMed
- 83. McAuley KA, Williams SM, Mann JI, Walker RJ, Lewis-Barned NJ, Temple LA, et al. Diagnosing insulin resistance in the general population. Diabetes Care 2001;24:460-4.ArticlePubMedPDF
- 84. Stumvoll M, Mitrakou A, Pimenta W, Jenssen T, Yki-Jarvinen H, Van Haeften T, et al. Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care 2000;23:295-301.ArticlePubMedPDF
- 85. Avignon A, Boegner C, Mariano-Goulart D, Colette C, Monnier L. Assessment of insulin sensitivity from plasma insulin and glucose in the fasting or post oral glucose-load state. Int J Obes Relat Metab Disord 1999;23:512-7.ArticlePubMedPDF
- 86. Gutt M, Davis CL, Spitzer SB, Llabre MM, Kumar M, Czarnecki EM, et al. Validation of the insulin sensitivity index (ISI(0,120)): comparison with other measures. Diabetes Res Clin Pract 2000;47:177-84.ArticlePubMed
- 87. Belfiore F, Iannello S, Volpicelli G. Insulin sensitivity indices calculated from basal and OGTT-induced insulin, glucose, and FFA levels. Mol Genet Metab 1998;63:134-41.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Polyphenol-Rich Extract of Fermented Chili Pepper Alleviates Insulin Resistance in HepG2 Cells via Regulating INSR, PTP1B, PPAR-γ, and AMPK Pathways
Tao Wang, Meiqi Li, Shengbao Cai, Linyan Zhou, Xiaosong Hu, Junjie Yi
Fermentation.2023; 9(2): 84. CrossRef - 2,3-Dihydrosorbicillin and chrysopanol stimulate insulin secretion in INS-1 cells
Dahae Lee, Jaekyung Kim, Sungyoul Choi, Jinwon Choi, Jin Woo Lee, Ki Sung Kang, Sang Hee Shim
Bioorganic & Medicinal Chemistry Letters.2023; 83: 129186. CrossRef - Insulin: A connection between pancreatic β cells and the hypothalamus
Brenda De la Cruz Concepción, Yaccil Adilene Flores Cortez, Martha Isela Barragán Bonilla, Juan Miguel Mendoza Bello, Monica Espinoza Rojo
World Journal of Diabetes.2023; 14(2): 76. CrossRef - The Metabolic Score for Insulin Resistance (METS-IR) Predicts Cardiovascular Disease and Its Subtypes in Patients with Hypertension and Obstructive Sleep Apnea
Wenbo Yang, Xintian Cai, Junli Hu, Wen Wen, Heizhati Mulalibieke, Xiaoguang Yao, Ling Yao, Qing Zhu, Jing Hong, Qin Luo, Shasha Liu, Nanfang Li
Clinical Epidemiology.2023; Volume 15: 177. CrossRef - Association between dietary patterns and biomarkers in connection with diabetes mellitus in adolescents: A systematic review
Bernardo Paz Barboza, Camila Tureck, Liliana Paula Bricarello, Mariane de Almeida Alves, Anabelle Retondario, Amanda de Moura Souza, Ricardo Fernandes, Francisco de Assis Guedes de Vasconcelos
Nutrition, Metabolism and Cardiovascular Diseases.2023; 33(4): 685. CrossRef - PNPLA3 rs738409 risk genotype decouples TyG index from HOMA2-IR and intrahepatic lipid content
Ákos Nádasdi, Viktor Gál, Tamás Masszi, Anikó Somogyi, Gábor Firneisz
Cardiovascular Diabetology.2023;[Epub] CrossRef - Ethnic Variability in Glucose and Insulin Response to Rice Among Healthy Overweight Adults: A Randomized Cross-Over Study
Amena Sadiya, Vidya Jakapure, Vijay Kumar
Diabetes, Metabolic Syndrome and Obesity.2023; Volume 16: 993. CrossRef - Familial partial lipodystrophy type 2 and obesity, two adipose tissue pathologies with different inflammatory profiles
Guillaume Treiber, Marie-Paule Gonthier, Alice Guilleux, Samir Medjane, Oriane Bonfanti, Muriel Cogne, Olivier Meilhac, Estelle Nobecourt
Diabetology & Metabolic Syndrome.2023;[Epub] CrossRef - Discriminant Model for Insulin Resistance in Type 2 Diabetic Patients
Erislandis López-Galán, Rafael Barrio-Deler, Manuel Alejandro Fernández-Fernández, Yaquelin Del Toro-Delgado, Isaac Enrique Peñuela-Puente, Miguel Enrique Sánchez-Hechavarría, Mario Eugenio Muñoz-Bustos, Gustavo Alejandro Muñoz-Bustos
Medicina.2023; 59(5): 839. CrossRef - Mechanisms of Oxidative Stress in Metabolic Syndrome
Sepiso K. Masenga, Lombe S. Kabwe, Martin Chakulya, Annet Kirabo
International Journal of Molecular Sciences.2023; 24(9): 7898. CrossRef - Estimated Glucose Disposal Rate Predicts Renal Progression in Type 2 Diabetes Mellitus: A Retrospective Cohort Study
Juan Peng, Aimei Li, Liangqingqing Yin, Qi Yang, Jinting Pan, Bin Yi
Journal of the Endocrine Society.2023;[Epub] CrossRef - Transfer RNA Mutation Associated with Type 2 Diabetes Mellitus
Fanny Rizki Rahmadanthi, Iman Permana Maksum
Biology.2023; 12(6): 871. CrossRef - Honokiol improves endothelial function in type 2 diabetic rats via alleviating oxidative stress and insulin resistance
An He, Huilin Yu, Yu Hu, Huiling Chen, Xiang Li, Jian Shen, Rongjuan Zhuang, Yi Chen, Bryan Richard Sasmita, Minghao Luo, Dingyi Lv
Biochemical and Biophysical Research Communications.2022; 600: 109. CrossRef - Relationship of Vitamin A and Thyroid Function in Individuals With Obesity and After Laparoscopic Sleeve Gastrectomy
Bingwei Ma, Peng Yang, Jingyang Gao, Lei Du, Chunjun Sheng, Taofeek Usman, Xingchun Wang, Shen Qu
Frontiers in Nutrition.2022;[Epub] CrossRef - Efficacy of Personalized Diabetes Self-care Using an Electronic Medical Record–Integrated Mobile App in Patients With Type 2 Diabetes: 6-Month Randomized Controlled Trial
Eun Young Lee, Seon-Ah Cha, Jae-Seung Yun, Sun-Young Lim, Jin-Hee Lee, Yu-Bae Ahn, Kun-Ho Yoon, Min Kyung Hyun, Seung-Hyun Ko
Journal of Medical Internet Research.2022; 24(7): e37430. CrossRef - Acyclovir alleviates insulin resistance via activating PKM1 in diabetic mice
Zhuozhou Hu, Jing Zhou, Liang Han, Xiangxiang Li, Chun Li, Tongyu Wu, Jingjing Liu, Wenyang Zhao, Jia Kang, Xinping Chen
Life Sciences.2022; 304: 120725. CrossRef - Identifying Glucose Metabolism Status in Nondiabetic Japanese Adults Using Machine Learning Model with Simple Questionnaire
Tomoki Uchida, Takeshi Kanamori, Takanori Teramoto, Yuji Nonaka, Hiroki Tanaka, Satoshi Nakamura, Norihito Murayama, Rajesh Kaluri
Computational and Mathematical Methods in Medicine.2022; 2022: 1. CrossRef - Elevated triglyceride-glucose (TyG) index predicts impaired islet β-cell function: A hospital-based cross-sectional study
Zi Chen, Jie Wen
Frontiers in Endocrinology.2022;[Epub] CrossRef - Maternal Glycaemic and Insulinemic Status and Newborn DNA Methylation: Findings in Women With Overweight and Obesity
Marion Lecorguillé, Fionnuala M McAuliffe, Patrick J Twomey, Karien Viljoen, John Mehegan, Cecily C Kelleher, Matthew Suderman, Catherine M Phillips
The Journal of Clinical Endocrinology & Metabolism.2022; 108(1): 85. CrossRef - Acanthosis Nigricans: Pointer of Endocrine Entities
Andreea-Maria Radu, Mara Carsote, Mihai Cristian Dumitrascu, Florica Sandru
Diagnostics.2022; 12(10): 2519. CrossRef - Relationship between Insulin Resistance Risk Scales and Non-Alcoholic Fatty Liver Disease and Liver Fibrosis Scales in 219,477 Spanish Workers
José Ignacio Ramírez-Manent, Emilio Martínez-Almoyna, Carlos López, Carla Busquets-Cortés, Hilda González San Miguel, Ángel Arturo López-González
Metabolites.2022; 12(11): 1093. CrossRef - Investigation of Insulin Secretion in Glucose Tolerance Test by the Intake of Novel Imeglimin (Twymeeg)
Hiroshi BANDO, Hiroko OGAWA, Hirohisa URASAKI, Shinji NAGAHIRO, Hiroko URASAKI, Miwako NAKANISHI, Osami WATANABE
Asploro Journal of Biomedical and Clinical Case Reports.2022; 5(3): 113. CrossRef - Evaluation and Screening of Hypoglycemic Activity of Total Ginsenosides GBE-5 Fraction From Panax Ginseng Berry Based on UHPLC–MS Metabolomics
Heyu Wang, Yu Tong, Anqi Wang, Ying Li, Bofan Lu, Hui Li, Lili Jiao, Wei Wu
Frontiers in Nutrition.2022;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite