Ubiquitin Ligases in Cholesterol Metabolism
Article information
Abstract
To maintain cholesterol homeostasis, the processes of cholesterol metabolism are regulated at multiple levels including transcription, translation, and enzymatic activity. Recently, the regulation of protein stability of some key players in cholesterol metabolism has been characterized. More and more ubiquitin ligases have been identified including gp78, Hrd1, TRC8, TEB4, Fbw7, and inducible degrader of low density lipoprotein receptor. Their working mechanisms and physiological functions are becoming revealed. Here, we summarize the structure, substrates and function of these ubiquitin ligases. Their potential application in drug discovery is also discussed.
INTRODUCTION
Cholesterol plays essential roles in mammalian cells. It is a key component of membrane and a precursor for the synthesis of steroid hormones and bile acids [1,2]. The cellular cholesterol homeostasis is achieved through balancing the following aspects: uptake by low density lipoprotein receptor (LDLR), de novo biosynthesis from acetyl-CoA, esterification with fatty acids, efflux mediated by ABC family transporters, and the secretion mediated by apolipoprotein B (ApoB) (Fig. 1). These pathways are regulated at both transcription and post-transcription levels. Sterol regulatory element binding protein (SREBP) and liver X receptor (LXR) are two major transcription factors controlling the expression of these genes. On the other hand, some key enzymes/factors are degraded through ubiquitin-proteasome pathway.
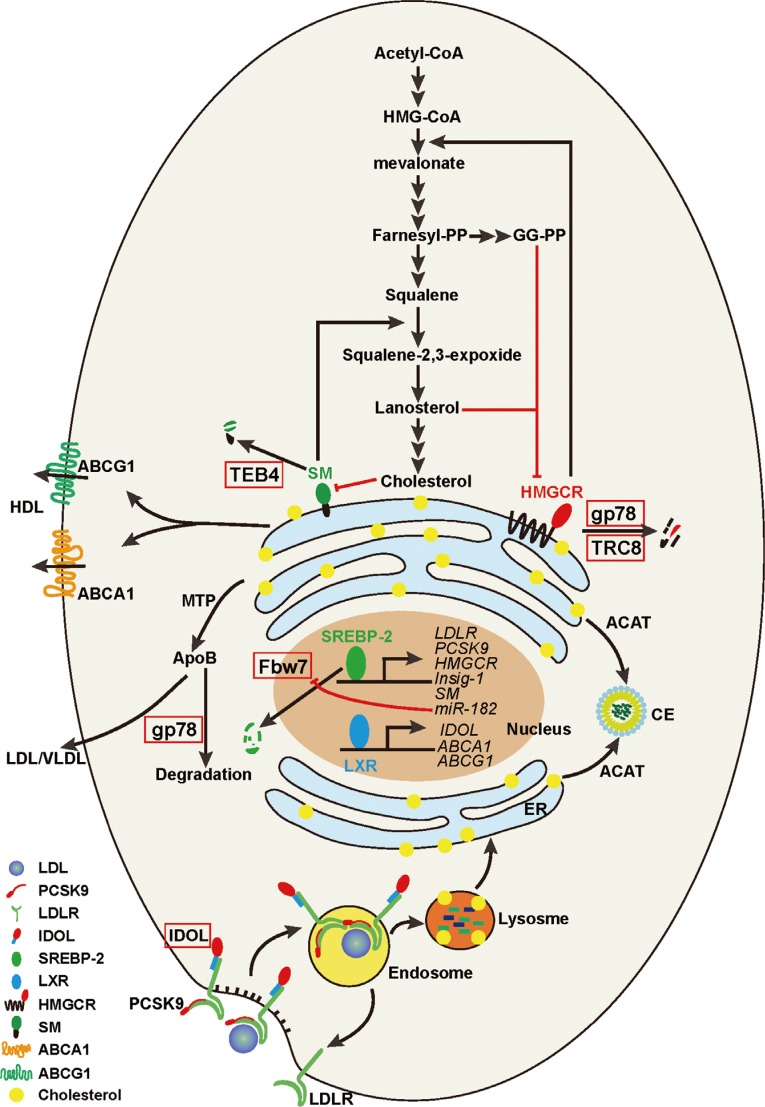
An illustration of cholesterol homeostasis in a typical mammalian cell. This diagram shows the cellular cholesterol metabolism contains at least four major routes: 1) the cholesterol de nove biosynthesis from acetyl-CoA in the endoplasmic reticulum (ER); 2) the low density lipoprotein (LDL) receptor-mediated endocytosis of LDL-derived cholesterol from plasma; 3) the efflux mediated by ABC family transporters such as ATP-binding cassette, sub-family A (ABC1), member 1 (ABCA1)/ATP-binding cassette, sub-family G (WHITE), member 1 (ABCG1) and the secretion mediated by apolipoprotein B (ApoB); and 4) cholesterol is esterified to cholesterol esters (CE) by acyl-coenzyme A:cholesterol acyltransferase (ACAT). See text for more details. HMG-CoA, 3-hydroxy-3-methyl-glutaryl-CoA; HDL, high density lipoprotein; TEB4, also called Membrane-Associated RING Finger Protein 6 (MRCH60); SM, squalene monoxygnease; HMGCR, HMG-CoA reductase; gp78, glycoprotein 78; TRC8, translocation in renal carcinoma on chromosome 8 protein; MTP, microsomal triglyceride transfer protein; SREBP-2, sterol regulatory element binding protein 2; Fbw7, F-box and WD repeat domain containing 7; LDLR, LDL receptor; PCSK9, proprotein convertase subtilisin/kexin type 9; Insig-1, insulin induced gene 1; IDOL, inducible degrader of LDLR; VLDL, very low density lipoprotein; LXR, liver X receptor.
Ubiquitin is a 76 amino acids (aa.). small protein that is conjugated to substrates for degradation. This process involves three enzymes-ubiquitin activating enzyme or E1, ubiquitin conjugating enzyme or E2, and ubiquitin ligase or E3. The ubiquitination is initiated by E1 that activates ubiquitin to conjugate to a cysteine residue in the E1 active site through the thiol group. Then the ubiquitin is transferred to the active site cysteine residue of second enzyme, E2. Finally, E3 catalyzes the ligation of ubiquitin with a lysine (or other) residue in the substrate [3].
The typical mammalian genome encodes only two E1s, but dozens of E2s and hundreds of E3s. Almost all known E3s can be classified into three ubiquitin ligase families-really interesting new gene (RING), homologous to E6AP carboxyl terminus (HECT), and U-box (UFD2 homology) proteins [3]. The diversity of E3s defines the substrate specificity. In this review, we will highlight the ubiquitin ligases (E3s) in regulation of key enzymes/factors involved in cellular cholesterol homeostasis (Table 1).
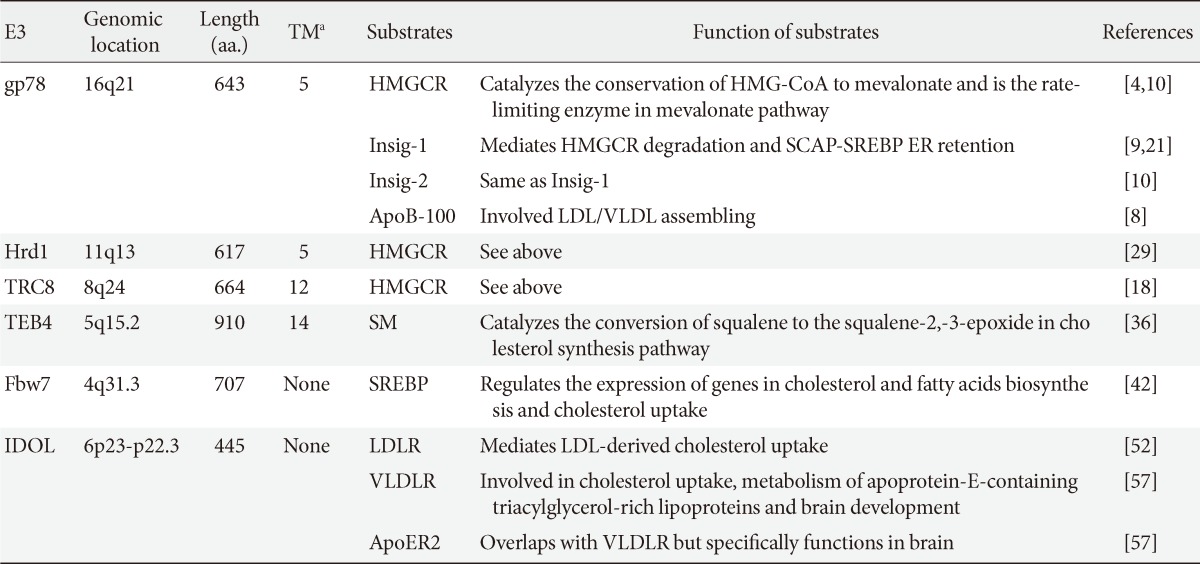
List of ubiquitin ligases disscussed in the text
gp78
Human glycoprotein 78 (gp78) is a 643 aa. protein which is composed of multiple domains. The NH2-terminal hydrophobic part (1-298 aa.) contains about five membrane span helixes. The following is a RING finger domain (340-382 aa.) sharing consensus sequences with other C3H2C3 RING finger containing proteins, and an oligomerization site (419-448 aa.), a coupling of ubiquitin to endoplasmic reticulum (ER) degradation (Cue) domain (456-497 aa.), an E2 UBE2G2 (Ubc7)-binding region (G2BR) (579-600 aa.), and a p97/VCP-interacting motif (VIM) (595-643 aa.) [4]. Additionally, there is an Ufd1 binding region (383-497 aa.) overlapped with Cue domain [5]. The fact that gp78 contains multiple conserved domains suggests that it is a multifaceted ubiquitin ligase and may be subjected to complicated regulations.
gp78 was originally identified as an autocrine motility factor receptor, a membrane glycoprotein of MW 78,000 from murine melanoma cells and mediated the tumor invasion and metastasis [6]. However, further studies defined gp78 as an ER-membrane anchored ubiquitin ligase [7]. It can promote the degradation of several misfolded proteins in the ER by recruiting ubiquitin-conjugating enzyme (E2) Ubc7/UBE2G2, Ufd1, and p97/VCP [4,5,7]. Later, several key proteins in cholesterol homeostasis including ApoB-100, insulin-induced gene 1 and 2 proteins (Insig-1/2, or Insigs), and 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) are found to be substrates of gp78 [4,8,9,10].
gp78 mediates the sterol-regulated ubiquitination of HMGCR
HMGCR is a rate-controlling enzyme of the mevalonate pathway which produces cholesterol and other isoprenoids (Fig. 1) [11]. HMGCR contains two domains: 1) the NH2-terminal-transmembrane domain which anchors the reductase on the ER-membrane, and 2) the COOH-catalytic domain which converts HMG-CoA to mevalonate. The expression of HMG-CR is transcriptionally regulated by SREBP in response to cellular sterols. Meanwhile, the protein level of reductase is also modulated by ubiquitin proteasome system which is a major negative feedback regulatory mechanism governing cholesterol biosynthesis [11].
High concentration of sterol, to be more precise, lanosterol, promotes the NH2-terminal transmembrane domain of HMGCR to interact with Insigs [12,13]. However, the Insigs themselves do not ubiquitinate HMGCR because they have no any ubiquitin ligase activity. Studies based on the permeabilized cell system show that sterol-dependent Insig binding results in recruitment of ubiquitin ligase [14]. This enzyme is then identified as gp78 by affinity purification coupled with tandem mass spectrometry [4]. gp78 binds Insig-1 constitutively in the ER membrane. When the cellular sterol level is high, the Insig-1/gp78 complex binds the transmembrane domain of HMGCR. Through the cascade reactions cooperated with E1, E2 (Ubc7), and E3 (gp78), as well as other cofactors, such as Ufd1 [5], the reductase was modified by Lys-48 linkage ubiquitin chains at the K248 (the dominant ubiquitination site) and K89 [15]. With assistance of at least two proteins associated with gp78, p97/VCP, and Aup1 [4,16], the ubiquitinated reductase was translocated to lipid droplet-associated ER membrane and dislocated from membrane into cytosol for proteasomal degradation [16,17]. This postubiquitination process can be promoted by geranylgeraniol (GG-OH) or its metabolically active geranylgeranyl-pyrophosphate (GG-PP) [15].
To further clarify the physiological effect of gp78 on lipid homeostasis, our group generated liver-specific gp78 knockout (L-gp78-/-) mice and has shown that the ablation of gp78 greatly increased the amount and activity of HMGCR in mice livers. Analysis from gp78-deficient primary hepatocytes indicated that the stability of HMGCR was increased through decreasing its sterol-stimulated ubiquitination and degradation [10]. These data demonstrate that gp78 is a physiological E3 regulating the stability of HMGCR.
Besides gp78, Jo and his colleagues [18] have found that another ubiquitin ligase translocation in renal carcinoma on chromosome 8 protein (TRC8) may also be involved in the regulation of reductase stability (discussed below).
gp78 mediates the degradation of Insig-1 and Insig-2
Human Insig-1 and Insig-2 proteins contain 277 and 225 aa., respectively. Both Insigs comprise six membrane spanning helices located in the ER. These two proteins are conserved even though they have a distinct NH2-terminal sequence [19,20]. On the one hand, both Insig-1 and Insig-2, can bind to SREBP cleavage activating protein (SCAP) and retain the SCAP/SREBP complex in the ER [19,20]. On the other hand, both Insigs can bridge the interaction between HMGCR and gp78, therefore lead to the ubiquitination of HMGCR by gp78 at high sterol condition [4,15,18].
Lee and Ye [21] have shown that Insig-1 has a rapid protein turnover rate in Chinese hamster ovary cells (CHO) and SV589 (a human fibroblast cell line) cells. The ubiquitination of Insig-1 is mediated by gp78 and regulated by sterols. Insig-1 is modified by gp78 at low sterol condition. High sterol promotes SCAP to bind Insig and gp78 is competed off, thereby stabilizing Insig-1 [9,22]. In sharp contrast to Insig-1, Insig-2 is quite stable in CHO and SV589 cells [21,22].
However, the protein level of Insig-2 is strikingly increased in gp78 deficient mice livers or primary hepatocytes. The Insig-1 level is moderately increased too [10]. Studies on constitutive gp78 knockout mice have found that the Insig-2 level in other tissues, such as brain and muscle does not alter (data not shown). Together, these results indicate that gp78 specifically promotes the degradation of Insig-2 in liver and white adipocytes [10].
In fact, it is a paradox that gp78 deficiency increases both the HMGCR and Insigs protein levels in mouse liver. This is because Insigs not only negatively regulates HMGCR posttranscriptionally but also inhibits the SREBPs processing through binding to SCAP [23]. These two outcomes are contradictory regarding cholesterol biosynthesis. Studies from L-gp78-/- mice have shown that the biosynthesis of cholesterol and fatty acids is decreased in gp78-deficient mouse liver [10]. This means the Insig-SCAP-SREBP axis dominates even though HMGCR is elevated. Moreover, the L-gp78-/- mice were also protected from diet-/age-induced obesity through increasing thermogenesis in brown adipocytes [10]. This study suggests that gp78 inhibitors may be used to treat hyperlipidemia and obesity.
gp78 mediated the degradation of ApoB-100 and secretion of LDL/VLDL particles
Liang and his colleagues [8] have identified another substrate of gp78 involved in cholesterol metabolism, ApoB-100. ApoB-100 is an essential protein component of very low density lipoproteins (VLDL) and low density lipoproteins (LDL) and plays critical roles in plasma cholesterol transportation (Fig. 1). There is considerable evidence showing that overproduction of ApoB is a common feature of dyslipidemia. ApoB-100 is one of committed secretory proteins at normal conditions. However, when the cellular lipid availability is limited, such as the new synthesized core lipids (triglyceride, cholesterol ester) or microsomal triglyceride transfer protein activity is decreased, the nascent ApoB-100 is subjected to ER-associated degradation mediated by gp78. When gp78 is overexpressed, the ubiquitination and degradation though 26S proteasome of apoB-100 is dramatically increased, and the secreted mature ApoB-100 is decreased [8]. When gp78 is knocked down, the secretion of ApoB-100 and the assembly of VLDL are increased in HepG2 cells [24]. The retrotranslocation of ApoB-100 also requires p97/VCP, similar to HMGCR [4,24].
In addition to HMGCR, Insig-1, Insig-2, as well as ApoB-100, a series of other proteins are identified as substrates of gp78. These proteins are CYP3A1, CYP2E1, deletion of phenylalanine 508 of cystic fibrosis transmembrane conductance regulator (CFTRδF508), ATZ, cholera toxin (CT), KAI1, mutant huntingtin (htt), neuroserpin, ataxin-3, and SOD1 [25]. Therefore, the gp78 knockout mice can serve as a valuable tool to dissect its multifaceted roles that may link the cholesterol metabolism to other physiological or pathological processes.
Hrd1
Human Hrd1 (also known as Syvn1) is an ER membrane-resident ubiquitin ligase which is comprised of 617 aa. The full-length proteins of Hrd1 and gp78 exhibit 28% similarity and 18% identity. Their RING finger regions show 69% similarity and 54% identity [26]. Similar with gp78, Hrd1 also uses Ubc7 as an E2 [7,26]. It has been reported that Hrd1 and gp78 have shared many substrates such as CD3-δ, CT as well as CFTRδF508 [7,26,27,28]. The ortholog of Hrd1 in yeast is hrd1p/Der3p that mediates the ubiquitination of Hmg2p, one of the yeast isozymes of HMGR [29]. This raises the possibility that Hrd1 may also mediate the turnover of HMGCR in mammalian cells. However, Hrd1 does not interact with Inisg-1 [4]. Moreover, RNAi-mediated gene silencing of Hrd1 did not block the sterol-stimulated ubiquitination of HMGCR in animal cells [4]. Whereas, the Hrd1 was indeed shown to control the basal but not sterol-regulated degradation of reductase in mammalian cells [26]. Therefore, the physiological role of Hrd1 needs to be further investigated.
TRC8
Human TRC8 is a multi-pass membrane protein located in the ER membrane. This protein comprises at least two distinct domains, the N-terminal multiple transmembrane domain containing predicted sterol sensing domain (SSD) and C-terminal RING domain with ubiquitin ligase activity [30,31]. This protein was originally identified from a family with hereditary renal cell carcinoma and thyroid cancer with which is disrupted by a constitutional translocation [31].
TRC8 contains a conserved SSD which is also present in other proteins including HMGCR, SCAP, Niemann-Pick disease, type C1 (NPC1), NPC1-like 1 (NPC1L1), and patched (a Hedgehog receptor) [31,32]. Furthermore, TRC8 can interact with both Insig-1 and Insig-2 proteins. RNAi study in SV-589 cells demonstrates that knockdown of TRC8 combined with gp78 can dramatically decrease the sterol-regulated ubiquitination as well as degradation of HMGCR [18]. This study indicates that both gp78 and TRC8 are involved in the sterol-accelerated ubiquitination of HMGCR in CHO-7 and SV-589 cells.
Interestingly, gp78 and TRC8 seemed dispensable for the degradation of HMGCR in mouse embryo fibroblasts (MEF) cells [33], suggesting that there are other ubiquitin ligases, at least in MEF cells, are required.
In addition, TRC8 binds both Insig-1 and Insig-2 [18]. It has been reported that TRC8 can ubiquitinate Insigs [30]. Similar to gp78, TRC8 may play opposite roles in cholesterol biosynthesis. It will be interesting to measure the metabolic changes in TRC8-deficient mice and compare them with gp78 knockout mice.
TEB4
Human TEB4 is a 910 aa. ER membrane-resident ubiquitin ligase which contains a conserved C4HC3 RING finger at the N-terminus and 14 predicted transmembrane helixes at C-terminus [34,35]. It has been shown that TEB4 interacts with ubiquitin conjugating enzyme Ubc7 through its RING domain and catalyzes K48-specific ubiquitin-ubiquitin linkage [35].
The homology of TEB4 in yeast is Doa10. Foresti and his colleagues [36] identified Erg1, which is the homologue of squalene monooxygenase (SM) in mammals, as a substrate of Doa10 in yeast. The Doa10 dependent degradation of Erg1 is regulated by lanosterol. High level of lanosterol promotes the degradation of Erg1. But in mammalian cells it is cholesterol that stimulates the degradation of SM mediated by TEB4 [36]. SM is a microsomal flavin monooxygenase that catalyzes the first oxygenation step during cholesterol synthesis, the conversion of squalene to the squalene-2,-3-epoxide (Fig. 1) [37]. As one of the target genes of SREBP-2, the transcription of SM is regulated by sterols [37]. Beyond the transcriptional regulation, the stability of SM protein is controlled by sterols too. Gill and his colleagues [38] have shown that the SM protein level is negatively regulated by cholesterol in mammalian cells. When the cholesterol, but not 24, 25-dihydrolanosterol, or side-chain oxysterols such as 27-hydroxycholesterol is present, the SM ubiquitinated by TEB4 [36,38]. The NH2-terminal domain (1-100 aa.) of SM is necessary and sufficient for the cholesterol-dependent degradation [38]. The regulated degradation of SM by TEB4 provides a possibility to treat hyperlipidemia by targeting TEB4.
Besides SM, TEB4 can ubiquitinate type 2 deiodinase (D2), which activates the prohormone thyroxine (T4) to the biologically active T3 molecule [39].
Fbw7
SKP1-cullin-1-F-box complex ubiquitin ligases (SCFs) are the largest and most versatile class of E3s. This complex contains Skp1, Cul1, Rbx1, and one of F-box proteins in mammalian cells, in which the F-box protein determines the recognition of substrates by binding to phosphorylated Ser and Thr residues of specific substrates [40]. F-box and WD repeat domain containing 7 (Fbw7 or Fbxw7), which belongs to Fbw family of F-box proteins, is identified as a tumor suppressor because its mutation has strong correlation with many cancers including breast, endometrial, ovarian, and colon cancer. This correlation can be partially explained by the fact that Fbw7 targets the substrates such as phosphorylated cyclin E, c-myc, as well as c-Jun for degradation [41]. Recent studies have identified other substrates of Fbw7, including SREBPs, C/EBP-α, and MCL1. They are involved in various biological processes including lipid metabolism, proliferation, and differentiation [42,43,44]. In this section, we will focus on the effect of Fbw7 on lipid hemostasis through modulating SREBPs stability directly.
SREBPs belong to basic helix-loop-helix-leucine zipper (bHLH-Zip) family of transcription factors. The nascent SREBPs are targeted to the ER membrane by the hairpin fashion without any transcription activity because they are not available for their target genes located in the nuclei. When cellular sterol is low, the SREBP precursors are translocated from ER to Golgi to be proteolytic processed by proteases. Then the NH2-terminal bHLH-Zip domain with the full transcription activity is then released from the membrane to reach the nucleus and acts as a transcription factor to active the genes, which are responsible for cholesterol and fatty acids biosynthesis and LDL uptake [45]. There are three SREBP proteins (designed SREBP-1a, SREBP-1c, and SREBP-2) from two srebp genes (designed srebp1 and srebp2) [45].
A dozen of years ago, Hirano and his colleagues [46] have found that the nuclear fragments of SREBPs were degraded through ubiquitin-proteasome system. Moreover, the stability of these proteins was regulated by acetylation [47]. Sundqvist and colleagues [42] have identified Fbw7, one member of SCFs E3 families, can interact with SREBPs and facilitates their ubiquitination and degradation dependent on the phosphorylation on Ser/Thr residues mediated by GSK-3. When disruption of Fbw7 in human colon carcinoma cells HCT116, the nuclear forms of SREBPs proteins were increased and as a consequent, the synthesis of cholesterol and fatty acids, as well as the LDL uptake were also increased dramatically [42]. These phenotypes were confirmed in mice with liver-specific null mutations of Fbw7 which exhibited hepatomegaly and steatohepatitis with massive deposition of triglyceride [48]. This means that Fbw7 plays pivotal roles in lipogenesis dominantly mediated by SREBP pathway. Additionally, Fbw7 is negatively regulated by microRNA miR-182 reciprocally, which was activated by SREBP-2 directly in mouse [49].Therefore, Fbw7-SREBP-miR-182 axis providers a regulatory loop for intracellular lipid homeostasis.
IDOL
Inducible degrader of LDLR (IDOL) was originally cloned as myosin regulatory light chain interacting protein (MYLIP) encoding 445 aa. with N-terminal FERM (F for 4.1 protein, E for ezrin, R for radixin, and M for moesin) [50] and C-terminal RING domains [51]. IDOL moderates the degradation of LDLR and requires the E2 enzymes UBE2D [52,53].
Mutation of LDLR causes human familial hypercholesterolemia, a rare autosomal dominant genetic disease characterized by dramatically elevated plasma cholesterol levels [54]. The transcription of LDLR gene is regulated primarily by SREBP in a sterol responsive manner. Recent studies have found that the LDLR is also regulation at posttranscriptional level, such as proprotein convertase subtilisin/kexin type 9 (PCSK9)-mediated degradation of LDLR in lysosome [55]. PCSK9 is the ninth member of proprotein convertase family, which is synthesized as a ~74 kDa soluble zymogen (proPCSK9) in the ER where it undergoes autocatalytic processing to release a processed enzyme of about 60 kDa to secrete to out of cells. It binds the extracellular domain of LDLR (Fig. 1). PCSK9 binding leads to lysosome degradation of LDLR rather than recycling from endosome to cell surface (Fig. 1).
Beyond the lysosomal degradation promoted by PCSK9, Zelcer et al. [52] have identified IDOL as a new posttranscriptional regulator of LDLR (Fig. 1). They observed that activation of LXR can decrease the abundance of LDLR without changing its mRNA level and subsequently inhibited uptake of LDL in different cells [52]. This means the regulation of LDLR mediated by LXR was attributed to posttranscriptional level. Later, gene expression microarrays were conducted and identified IDOL as a potential candidate in this process [52]. The gain of function experiments in various cell lines and mice demonstrated that IDOL can increase the plasma cholesterol level. These effects were achieved by ubiquitination and degradation of LDLR dependent on its cytosolic domain. Loss of function studies in different cells including embryonic stem cells (Idol-/- ES) have shown that the decrease or ablation of IDOL can elevate the LDLR protein level and promote LDL uptake. Moreover, in Idol-/- ES cells, the LDLR protein abundance was not responsive to LXR agonist any more [56]. These data suggested that the IDOL was a primary ubiquitin ligase to modulate LDLR. However, the expression level of Idol in liver is relatively low, and it is not regulated by LXR [52]. Rather, the LXR-IDOL pathway seems to be more active in peripheral cells such as macrophages, small intestine and adrenals [52]. The physiological significance should be dissected in more detail.
Additionally, IDOL can mediate the ubiquitination of other two LDLR family members, VLDL receptor and ApoE receptor 2 with a conserved mechanism [57]. These suggest that the IDOL is an import mediator of LDL and VLDL at posttranscriptional level.
CONCLUSIONS
In conclusion, ubiquitin ligase-mediated degradation of key proteins plays essential roles for cholesterol homeostasis. Thus, it provides alternative ways to develop drugs to lower cholesterol level. The three-dimensional structure of ubiquitin ligase-substrate complexes may be helpful. The specificity of inhibitors/activators should be considered too. Meanwhile, as most of the E3s related to cholesterol metabolism are membrane proteins, it may be difficult and special techniques may be required. On the other hand, the deubiquitinating enzymes (DUBs) that counteract ubiquitin ligases can regulate the cellular protein homeostasis [58]. Further studies should be also required to identify which DUBs are involved in the cholesterol metabolism and to explore the underlying mechanisms.
Notes
No potential conflict of interest relevant to this article was reported.