Hyperinsulinemia in Obesity, Inflammation, and Cancer
Article information

Abstract
The relative insufficiency of insulin secretion and/or insulin action causes diabetes. However, obesity and type 2 diabetes mellitus can be associated with an absolute increase in circulating insulin, a state known as hyperinsulinemia. Studies are beginning to elucidate the cause-effect relationships between hyperinsulinemia and numerous consequences of metabolic dysfunctions. Here, we review recent evidence demonstrating that hyperinsulinemia may play a role in inflammation, aging and development of cancers. In this review, we will focus on the consequences and mechanisms of excess insulin production and action, placing recent findings that have challenged dogma in the context of the existing body of literature. Where relevant, we elaborate on the role of specific signal transduction components in the actions of insulin and consequences of chronic hyperinsulinemia. By discussing the involvement of hyperinsulinemia in various metabolic and other chronic diseases, we may identify more effective therapeutics or lifestyle interventions for preventing or treating obesity, diabetes and cancer. We also seek to identify pertinent questions that are ripe for future investigation.

INTRODUCTION
Insulin is a peptide hormone secreted by pancreatic β-cells to regulate glucose homeostasis. Since its discovery in Canada in 1921 [1], insulin has saved millions of lives. However, in recent years, many studies have demonstrated that excess insulin (also known as hyperinsulinemia) might play a complex role in multiple diseases. In this review, we will focus on the cause-effect relationships between hyperinsulinemia, aging, obesity, inflammation, and cancers. As several complementary reviews have extensively discussed the importance of hyperinsulinemia in obesity [2-4] and cancer [5] separately, we will focus on expanding and integrating these concepts together. We will also discuss the potential molecular mechanisms mediating the effects of hyperinsulinemia on these disorders. We will discuss the limitations of the current evidence base and areas for future study. We hope that by targeting hyperinsulinemia, we may develop new therapeutic strategies for the prevention and/or treatment of obesity, diabetes, and cancers.
INSULIN AND INSULIN SIGNALING MECHANISMS IN THE CONTROL OF METABOLISM AND LIFESPAN
In order to better understand the (patho)physiological effects of insulin, we must first consider its evolution. Insulin and its related peptides are ancient, evolutionarily conserved, anabolic neurohormones [6-9]. Humans have one INS gene, two insulin-like growth factor genes (IGF1, IGF2), several insulin-like peptides, and relaxins [10-13]. Mice have two insulin genes. Mice lacking either Ins1 or Ins2 have normal glucose, suggesting a genetic compensation [14,15], but loss of both insulin genes is lethal [15,16]. Ins1 originated from a reverse-transcribed and partially processed Ins2 mRNA transposition [17] and is restricted to β-cells where it contributes 1/3rd of insulin synthesized and secreted into circulation [18]. Ins2 is the ancestral gene that produces 2/3 of the insulin mRNA in islets [19,20], as well as trace amounts in the thymus [21] and brain [20]. Using mice as a genetically tractable model for mammalian insulin physiology has provided insight into obesity, diabetes, cancer, and other aging-associated diseases where changes in insulin levels and signaling are implicated.
Insulin and IGF have many similarities but also have key differences in their function and downstream signaling mechanisms. Insulin is recognized for its acute metabolic roles, whereas the IGFs are more commonly studied in association with growth, aging, and cancer [22]. Emerging evidence points to important ‘growth factor’ roles of insulin [20,23-32], as well as ‘metabolic’ roles of IGF1/IGF2 [33,34]. A conserved signaling pathway is largely shared between insulin and IGFs; thus, it is difficult to clearly separate the effects of insulin and IGF1/IGF2. Unfortunately, the majority of studies in the literature, especially those reporting in vitro cell culture experiments, have used insulin concentrations that are many times higher than those found physiologically, confounding interpretation of these studies and clouding the field. Insulin binds to its receptor tyrosine kinase, insulin receptor (INSR) with an IC50 of 0.89 nM and to the growth factor 1 receptor (IGF1R) with an IC50 of 30 to 400 nM [35-37]. INSR/IGF1R can form heterodimer hybrid receptors that have a higher affinity for IGF1 than insulin [38-40]. Binding to the INSR and related receptors leads to tyrosine kinase autophosphorylation and binding of adapter proteins, called insulin receptor substrates (IRS1, IRS2, IRS3, IRS4), which attract the lipid kinase, phosphoinositide 3-kinase (PI3K). Phosphorylation of PI(4,5)P2 to PI(3,4,5)P3 by PI3K activates the PH-domain-containing kinase 3-phosphoinositide dependent protein kinase 1 (PDPK1), which in turn phosphorylates serine/threonine kinase AKT serine/threonine kinase 1 (AKT) [41]. Insulin-mediated activation of AKT leads to the phosphorylation of multiple signaling intermediates including the forkhead family box O (FOXO) family of transcription factors, glycogen synthase kinase 3 (GSK3), and the mammalian target of rapamycin complex 1 (mTORC1) complex [41,42]. Insulin signaling can also activate the Ras-family GTPase (Ras)–mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) phosphorylation cascade via the adapter proteins SHC-transforming protein 1 (SHC1), growth factor receptor-bound protein 2 (GRB2), and son of sevenless (SOS) [41]. There are multiple levels of cross talk between the major ‘arms’ of insulin signaling, as well as cross talk to other signaling networks [38-40,43]. When investigated individually, virtually all aspects of the extended insulin signaling network have been linked to diabetes when strongly suppressed, to longevity when mildly suppressed, and to cancer when overactive [31,38,44-47].
We have learned much about the role of insulin and insulin signaling in longevity by studying relatively simple model organisms. INSR or IRS mutant flies live 50% longer than wildtype flies [48,49]. Mice lacking IRS1 have increased lifespan compared to wildtype controls [50,51]. Studies in humans have linked insulin and IGF1 to longevity [9], with lower levels of these factors associated with longer life [52,53]. Mice with heterozygous loss of Igf1r were reported to have increased longevity [54], but this has been questioned [55]. Reducing circulating IGF1 by liver-specific gene ablation in female mice increases mean lifespan, but not maximum lifespan [56]. These observations and others demonstrate that IGF1 plays important roles in longevity [57], but also suggest that factors other than IGF1, such as insulin, may be important in some contexts. Mice lacking adipose tissue Insr are leaner and live longer than controls [58-60], but Insr loss also impacts IGF signaling via hybrid receptors [24,26,61]. Because the insulin and IGF ligands can (promiscuously) target receptor hetero dimers and each other’s homodimers, the receptor ligands must be manipulated directly to understand insulin’s role in metabolism and lifespan. Indeed, we found genetically reduced insulin production extended lifespan in mice [44]. In liver, gene networks related to cellular metabolism, circadian rhythm, proteostasis, and cell-cycle progression were altered in mice with reduced insulin [44]. Thus, insulin/insulin signaling is a positive regulator of energy storage and is a negative regulator of longevity in all animals studied to date (Fig. 1) [9,54,57,62,63]. Manipulations that reduce plasma glucose levels, and thereby circulating insulin, also extend lifespan [64], but it remains unclear whether the glucose effects are independent of insulin. Emerging evidence links hyperinsulinemia and/or hyperglycemia to mortality after infections, including severe acute respiratory syndrome coronavirus 2 [65,66], but these concepts also require further mechanistic studies to determine causality.
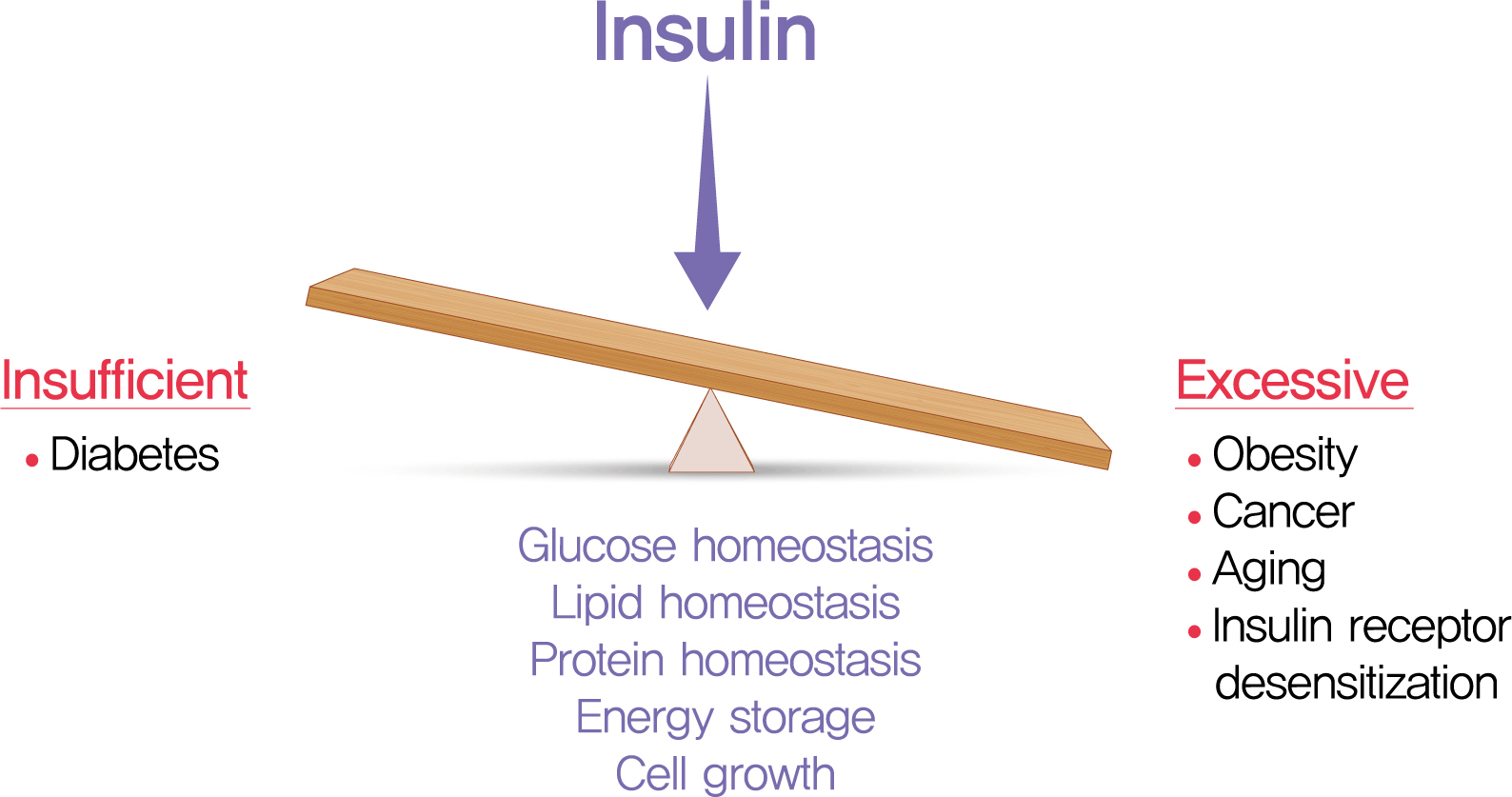
Trade-offs with insufficient and excessive circulating insulin levels.
"Updated on 30 July 2021"
HYPERINSULINEMIA PLAYS A CAUSAL ROLE IN OBESITY
The range of circulating insulin is relatively broad in healthy individuals and peaks approximately 30 minutes after glucose administration, remaining elevated above baseline for over 2 hours. Studies that tracked insulin over 24 hours found that fasting insulin averaged 60 pmol/L while post-meal insulin peaked at 420 pmol/L [67]. In individuals with obesity, fasting insulin was 140 pmol/L while post-meal insulin reached 840 pmol/L [67]. Fasting insulin of >85 pmol (12.2 mIU/L) has been proposed as a cutoff to define hyperinsulinemia and is sufficient to mark the pathological state of metabolic syndrome [68]. In all conditions, insulin levels are thought to be at least approximately 10 times higher in the pre-hepatic portal circulation and in the pancreas [69,70]. Conceptually, hyperinsulinemia can be defined as excess insulin relative to what is required to maintain normal glucose. Hyperinsulinemia can manifest as an elevation in basal/fasting circulating insulin and/or as a potentiation of post-prandial insulin secretion [71,72], and can result from insulin hypersecretion or reduced systemic insulin clearance, or both [73-76]. It is critical to understand which type of hyperinsulinemia is being studies when examining correlations with and causal effects on various (patho)physiological parameters.
Relative insulin insufficiency defines diabetes, but long before diagnosis of type 2 diabetes mellitus (T2DM), excess insulin predicts the people who will progress to disease [77,78]. The coincidental timing of hyperinsulinemia, insulin resistance, and obesity has led to ‘chicken and egg’ questions and extensive debate [2-4,79]. Conventional dogma places excess adiposity and insulin resistance as initial causes, with hyperinsulinemia as a consequence. The commonly accepted model holds that obesity causes hyperinsulinemia via insulin resistance. The insulin resistance is thought to arise primarily from the spill over of lipids into liver, muscle, and other tissue (i.e., ectopic lipid deposition), from adipose tissue that has exceeded its capacity for storage [4,41,80]. The obesity-induced insulin resistance is then thought to lead to hyperglycemia which then drives the pancreatic β-cells to secrete more insulin to maintain glucose homeostasis, causing hyperinsulinemia. However, clinical observations cast doubt that this paradigm is applicable in all cases, especially when hyperglycemia is often one of the last clinical features in the progression from obesity to T2DM and when insulin resistance is circularly defined as unexplained hyperinsulinemia [81]. Basal hyperinsulinemia has been documented to occur prior to insulin resistance, obesity and/or hyperglycemia [24,71,82-92]. Analysis of 1,168 non-diabetic adolescents and adults found that the upper tertile of insulin hypersecreting individuals showed significantly increased fat mass, worse lipid profile and impaired glucose tolerance independent of clamp-measured insulin resistance, compared to the lower tertiles [93]. Obesity is associated with increased basal and post-prandial insulin secretion even in subjects without insulin resistance [94]. Moreover, injected insulin is sufficient to induce insulin resistance in humans, compared with glycemia matched controls [95]. Epidemiological studies show that children with hyperinsulinemia had an increased risk of developing obesity later in life [82,92]. These observations cast doubt on the primacy of classically-defined insulin resistance in the pathogenesis of obesity and T2DM. From an endocrinological perspective, hormones are known to desensitize their receptors and post-receptor signaling processes when present in excess. Genetic downregulation of insulin production illustrated that hyperinsulinemia is a cause, and not just the consequence, of insulin resistance and elevated fasting glucose in old age [44]. Revisiting the concept of insulin resistance is beyond the scope of this review, but will be the subject of a forthcoming article.
The human genetics of obesity are complex but provide insight into cause-and-effect and the role of specific components of insulin signaling. Genome-wide association study (GWAS) studies have shown that the vast majority of the common variation underlying body mass index (BMI) is related to food intake centers in the brain [96]. FTO (FTO alpha-ketoglutarate dependent dioxygenase) is associated with hyperinsulinemia, among many other possible mechanisms [97]. INSR and other components of the insulin signaling pathway are associated with lipid and adiposity-related traits (www.ebi.ac.uk/gwas). Moreover, the INS locus itself is associated with anthropometric traits including height, birth weight, fat-free mass, and waist-hip ratio, in addition to both type 1 diabetes mellitus (T1DM) and T2DM (www.ebi.ac.uk/gwas). The INS locus is complex, and how these polymorphisms affect insulin secretion remain unclear, but are beginning to be investigated. Mendelian randomization studies support a causal role for genetically driven glucose-stimulated hyperinsulinemia in obesity [98], although studies also support a causal contribution of BMI to fasting insulin and insulin resistance [99]. It is likely that there is bi-directional causality and the results depend on which single-nucleotide polymorphisms and which anthropomorphic traits are chosen for analysis. Studies of rare human genetic conditions also support a causal role for insulin action in obesity. For example, increased global insulin sensitivity in humans with phosphatase and tensin homolog (PTEN) mutations causes obesity [100], whereas humans with INSR loss-of-function mutations (e.g., Donohue syndrome) have intrauterine growth restriction and reduced subcutaneous fat [101]. Together, these studies point the possibility that genetic variation in insulin and/or insulin signaling contributes to human obesity.
Multiple environmental and inherited factors can drive insulin hypersecretion and reducing these burdens has been shown to prevent or ameliorate disease. Thomas et al. [92] and Corkey [102] proposed environmental/diet factors that can drive primary hyperinsulinemia. Reducing fasting insulin with low carbohydrate diets, caloric restriction, or time-restricted feeding improves insulin sensitivity in mice and humans and may lead to T2DM remission [103-106]. Bariatric surgery simultaneously corrects hyperinsulinemia and diabetes, independent of weight loss, insulin sensitivity, or glucose [3]. However, even clinical studies with sensitive measures are often correlative, making direct experimental manipulation of insulin the most robust way to test the hypothesis that hyperinsulinemia drives chronic diseases, including diabetes, obesity, and even cancer. Indeed, blocking insulin secretion with diazoxide or octreotide causes weight loss in both rat model and human studies, supporting a causal role in obesity for excess insulin [107-112]. Clinical trials have shown that prolonged elevation of exogenous long-acting insulin analogues causes weight gain [113]. Collectively, there is strong clinical evidence in humans, both loss-of-function and gain-of-function studies, that support a primary, causal role for hyperinsulinemia in obesity and associated traits [2,71,79,92,102].
Complementing clinical studies, engineered mouse models provide genetic certainty that manipulation of insulin production causes changes in body weight and adiposity [2]. In an initial study, Mehran et al. [20] found that mice with only one allele of Ins1, which significantly reduced fasting insulin levels, were protected from high-fat diet-induced obesity and adipocyte hypertrophy compared to control mice with two alleles of Ins1 in an Ins2-null genetic background. Energy expenditure was increased, but there was no difference in measured food intake [20]. Follow-up studies identified upregulation of oxidative phosphorylation complex proteins and uncoupling protein 1 in white adipose tissue [114]. Templeman et al. [30,115] made similar observations by modulating Ins2 gene dosage in mice without Ins1. When experimental Ins1−/−;Ins2+/− mice were implanted with insulin releasing pumps, body weight, white adipose tissue mass and adipose tissue hypertrophy were partially restored [30]. D’Souza et al. [116] showed that obesity driven by leptin deficiency also requires hyperinsulinemia. Page et al. [117] used a conditional Ins2 partial ablation and the resulting modest reduction in insulin secretion to induce weight loss in mice that were already obese. In all of these experiments, changes in circulating insulin that were too small to adversely affect glucose homeostasis were nonetheless able to significantly alter adiposity. Our studies agree with work from other groups showing that mouse models with reduced insulin secretion are protected from diet-induced obesity [118] and that Insr deficiency in adipose tissue prevents diet- and age-induced obesity [26,59,119,120]. Multiple lines of evidence demonstrate that maximal adipocyte hyperplasia and hypertrophy require fully intact insulin action.
The biochemical rationale for insulin-driven obesity and insulin resistance are established. Insulin is a robust stimulator of lipid transport into adipocytes, adipocyte differentiation and a potent inhibitor of lipolysis in adipose tissue [121]. The specific mechanisms by which hyperinsulinemia affects adiposity remain understudied. One of the main effects of insulin on adipocytes is to promote lipogenesis through stimulation of fatty acid uptake and triglyceride synthesis [2,122,123]. Insulin can also inhibit lipolysis and induce the expression of transcription factors such as CCAAT/enhancer-binding protein β to modulate lipid uptake and storage [2,124,125]. More acute, inducible reduction of insulin production in adult mice had a specific effect on visceral adipose tissue and a reduction in the protein abundance of the human lipodystrophy gene, caveolae associated protein 1 (Cavin1) [117]. Therefore, excess insulin signaling in adipocytes can lead to excess fat accumulation and to obesity.
Insulin may also regulate peripheral lipid metabolism through the brain [119,124,125]; however, the actions of central insulin signaling on food intake and body weight remain controversial. For instance, insulin administration by intracerebroventricular injection increased fat mass and adipocyte size in some studies [26,119], but decreased food intake and body weight in others [126-130]. Moreover, some researchers observed that hypothalamic Insr deletion was accompanied by hyperphagia and fat mass increase [126,131,132], while some found the deletion caused a reduction in white adipose tissue mass [130]. The reasons for these inconsistencies are unclear. Likely, the direction of the effects depends on the specific brain regions in which insulin signaling is inhibited or activated [124,126].
EFFECTS OF HYPERINSULINEMIA ON INFLAMMATION
Inflammation is associated with metabolic dysfunction such as obesity and insulin resistance [133]. Several studies suggest that obesity-associated, chronic, low-grade inflammation can cause insulin resistance in metabolic tissues like liver, muscle, and adipose tissues [134-136]. However, insulin resistance can also precede inflammation [137], can occur independently of inflammation [138], and anti-inflammatory treatments have not been shown to cause weight loss in humans. The root cause and the ultimate consequences of inflammation in context of metabolic health remains to be fully understood. Nevertheless, there is a strong association between hyperinsulinemia and chronic low-grade inflammation. Modest hyperinsulinemia in experimental animals is sufficient to cause adipose tissue inflammation [139]. Mice with both chronically and acutely reduced insulin production exhibit a gene expression profile that suggests impaired innate immunity in adipose tissue [20,117]. Insulin infusion during euglycemic hyperinsulinemic clamp increases pro-inflammatory interleukin 6 (IL-6), tumor necrosis factor alpha (TNF-α), and monocyte chemoattractant protein-1 (MCP-1) in human serum and adipose tissue [140-143]. Thus, both human and mouse studies support the concept that hyperinsulinemia is necessary and sufficient to promote inflammation.
Hyperinsulinemia can contribute to tissue-specific inflammation in multiple ways. For example, we and others have shown that hyperinsulinemia leads to hypertrophic and unhealthy adipocytes [20,119,144,145], which can attract macrophages and other immune cells [146-149]. Hyperinsulinemia may also promote inflammation by direct effects on immune cells. Insr mRNA is expressed in multiple immune cell types, based on single-cell RNA sequencing data (Fig. 2A), suggesting that at least some effects of hyperinsulinemia may be direct. Previous studies have identified INSRs mRNA or protein in T-cells, B-cells, neutrophils, monocytes, macrophages, and natural killer (NK) cells [150-153]. INSR expression in murine T-cells increased with activation [151,152], suggesting a role in T-cell function. T-cells with specific Insr gene knockout exhibit reduced production of interferon gamma (IFNγ), IL-4, and IL-10 upon activation, as well as diminished cytotoxicity [151,153]. Insulin, acting through INSR, is also required for maximal T-cell proliferation and its loss leads to apoptosis sensitivity and defective differentiation toward the T helper cell 1 (Th1) and Th17 lineages [151,154]. Similarly, AKT inactivation limits proliferation and the production of IL-2, IL-4, and IFNγ in CD4+ T-cells [155]. Collectively, these studies suggest that insulin signaling regulates T-cell cytokine production and proliferation. T-cell activation requires glucose and glutamine metabolism [153,156-158]. Therefore, it is possible that insulin signaling controls activated T-cell functions through regulation of glucose metabolism (e.g., stimulating glycolysis and regulating glucose or amino acid transporters expression on T-cells). Although no studies have directly addressed how hyperinsulinemia affects T-cell function, it seems likely based on the above evidence that hyperinsulinemia would be pro-inflammatory. Obesity-induced hyperinsulinemia has been correlated with reduced regulatory T-cell proportions [153,159,160]. Both in vivo and in vitro studies showed that hyperinsulinemia reduced the regulatory T-cells’ ability to produce IL-10, which in turn impairs their ability to suppress TNF-α production in macrophages [153]. The effects of AKT/PI3K signaling on regulatory T-cell suppressive activity have been reported to be positive in some studies [155,161] and negative in others [155, 162]. Collectively, the available literature suggests that insulin and downstream signaling may have important and direct effects on both cytotoxic and regulatory T-cells.

Possible cellular targets of hyperinsulinemia in pancreatic cancer. (A) Violin plot showing insulin receptor (Insr) expression in pancreatic cells and immune cells. The data is based on single-cell gene expression analysis of mouse pancreas in Ptf1aCreER; LSL-KrasG12D; Ins1−/− genetic background generated using 10x Genomics. Background mRNA contamination was cleaned by R packages SoupX and the single-cell gene expression data was analyzed by Seurat 4.0 (Satija Lab). (B) Possible cellular targets of hyperinsulinemia in pancreatic cancer initiation and/or progression.
Insulin signaling also affects innate immune cells. Macrophages are responsible for detecting, destroying and engulfing pathogens and macrophages can polarize along a spectrum of M1 to M2 [163,164]. So called ‘M1 macrophages’ produce proinflammatory cytokines such as TNF-α, IL-6, and IL-1β which kill pathogens or tumor cells. ‘M2 macrophages’ produce antiinflammatory cytokines like IL-10 and transforming growth factor beta 1 (TGF-β), which heal wounds and resolve inflammation [136,163,164]. Macrophages are known to modulate both insulin secretion [165] and tissue insulin sensitivity [136,166,167]. Insulin signaling within macrophages has been extensively studied [168,169]. Macrophages express all components of insulin signaling cascade [170,171], and when Insr or Akt2 were deleted, macrophages shifted towards an anti-inflammatory phenotype with reduced IL-6, IL-1β, and TNF-α [170-173]. However, studies employing PI3K or AKT overexpression, PI3K knockout, or deletion of PTEN and Src homology 2-containing inositol 5ʹ-phosphatase have also implicated PI3K/AKT/mTOR signaling in M2 polarization [173-179]. These inconsistencies in M1/M2 polarization may be related to AKT and mTOR isoform specificity [173,174,180]. Moreover, given that PI3K/AKT/mTOR signaling can be stimulated by many ligands besides insulin, for example TGF-β, IL-10, and bone morphogenetic protein 7 [174,180], the role of this signaling cascade in macrophage function may be context dependent.
HYPERINSULINEMIA AND CANCER
Interest has grown recently around the possibility that excess insulin, a consequence of unhealthy diets and lifestyles, may have cancer-promoting effects. Insulin is a powerful mitogen and survival factor for virtually all cell types [23,32,181]. While insulin’s close relative IGF1 has been extensively studied for its roles in many cancers, studies on the potential role of insulin have lagged behind. Some epidemiological studies report increased cancer risk in patients receiving exogenous long-acting insulins as diabetes therapy [182-187], but these findings were controversial and subsequent studies have failed to find a consistent association [188-191]. Interest has now shifted towards the role of endogenous insulin in cancer risk. Despite correlative human data associating high levels of insulin with multiple types of cancer, the cause-and-effect relationship had not previously been established for any cancer [192-195].
Obesity and diabetes are risk factors for many different types of cancers [5,196-200]. In 2012, combined effects of high BMI and diabetes were responsible for 5.7% of cancer cases [201]. Broken down by cancer type, 16.5% to 18.8% of liver cancer, 13.1% of pancreatic cancer, 7.2% of breast cancer, 31.3% of endometrial cancer, 28.7% to 29.5% oesophageal cancer, and 18% to 21.3% of kidney cancers have been attributed to obesity and diabetes [201]. Compared to healthy people, patients with diabetes have 1.57-fold increased risk of death from cancer and more specifically, they were associated with about 2.34-fold, 8.47-fold, and 4.2-fold increased risk of deaths from pancreas, liver, and breast cancers, respectively [200,202]. Obesity is associated with 14% in colorectal and 35% increase in breast cancer-specific mortality, respectively [203,204]. Populations with diabetes and obesity are increasing rapidly [205-208] and the percentage of cancer cases attributable to obesity and diabetes increased 20% and 30%, respectively, between 1980 and 2002 [201]. Obesity and diabetes are also associated with increased cancer mortality [199,200,202,209-211]. There is an urgent need to identify the underlying mechanisms that link cancers to obesity and diabetes.
Hyperglycemia, hyperinsulinemia, increased inflammation and dyslipidemia usually accompany obesity and diabetes and those metabolic changes are considered as factors causing increased risks of cancers morbidity and mortality [5,209,211,212]. For instance, hyperglycemia may provide extra-glucose to cancer cells to maintain their rapid proliferation and meet the demand for biomass production. Hyperglycemia can cause overproduction of advanced glycation end-products and reactive oxygen species, which can cause DNA damages [209, 212]. Moreover, dysregulation of leptin and adiponectin production in adipocytes that is associated with obesity and diabetes can also affect cancer cells growth and survival [209,212]. Here, however, we are focused on hyperinsulinemia as a potential driver of increased cancer incidence and mortality. Previous scholarship has thoroughly reviewed other possible mechanisms [209,212].
Hyperinsulinemia is associated with cancer mortality independently of diabetes, obesity, and metabolic syndrome [213,214]. Insulin is a well-established growth factor for many cell types, including pancreatic cancer cells and their likely precursors [215]. In addition to pancreatic cancer (see below) [213, 216-219], people with hyperinsulinemia also had increased risk of breast [197,220-223], colorectal [196,224,225], prostate [226], endometrial [227,228], liver [198], and ovarian cancers [229], regardless of BMI. Hyperinsulinemia was associated with a 2-fold risk of cancer death [196,211,230,231]. This increase of cancer mortality is also observed in people with normal body weight if they had hyperinsulinemia [214]. Therefore, hyperinsulinemia is associated with increased risk of both cancer incidence and death. However, unlike hyperglycemia, there is no widely accepted insulin concentration to define hyperinsulinemia, so it is difficult to compare across studies. Nevertheless, the rationale to study the contribution of hyperinsulinemia to cancer is strong.
THE INSULIN RECEPTOR, INSULIN SIGNALING AND CANCER
Insr expression levels and isoform splicing have been investigated for their roles in cancer. There are two isoforms of INSR, INSR-A, and INSR-B. INSR-B includes the exon 11, which is not present in INSR-A [39,40]. INSR-A/INSR-A homodimers, INSR-A/IGF1R heterodimers, and INSR-B/IGF1R heterodimers can bind to insulin, IGF1, and IGF2; however, INSR-B/INSR-B homodimers can only bind to insulin when comparing the ligands at physiological concentrations [39,40]. INSR-A has a higher affinity to insulin than INSR-B. INSR-A is believed to mediate more of the mitogenic and anti-apoptotic effects of insulin while INSR-B is believed to carry more metabolic effects [39,40]. INSR-A is believed to be the isoform that is predominantly upregulated and overexpressed during cancer development [5,39,40,211]. The increase of INSR-A/INSR-B ratio has been found in breast [46,232-236], prostate [237-239], endometrial [240,241], colon [242], and lung cancers [242-244]. The mechanism of how cancer cells overexpress INSRs (especially INSR-A) is still unknown but some studies suggest that the transcription of Insr maybe dysregulated. Two transcription factors, high mobility group AT-Hook 1 and specificity protein 1 which positively regulate Insr expression, were found upregulated in some cancers [245-248]. On the other hand, tumor protein p53 (TP53), which is the negative regulator of Insr, frequently has loss-of-function mutation in cancers [249]. Some microRNAs that are known to be involved in regulating Insr and Igf1r are also dysregulated in certain cancers, although additional mechanistic studies are required [250,251]. Hyperinsulinemia can cause INSR internalization, which decreases the number of INSRs available on the cell membrane for binding the insulin ligand; however, some studies have found that breast cancer cells lose their sensitivity to hyperinsulinemia-induced INSR downregulation [46,234]. Furthermore, the increased INSR-A to INSR-B ratio was positively correlated with hyperinsulinemia, while the decreased INSR-A to INSR-B ratio was associated with low insulin levels after fasting or after bariatric surgery [40,252,253]. One of the mechanisms for this hyperinsulinemia-induced INSR-A upregulation could involve the insulin-induced degradation of splicing factor, which can shift INSR-A to INSR-B ratios [40,254]. Multiple drugs were designed to targeted IGF1R and INSR. However, the potential for insulin and IGF1 to bind both receptors complicates results from clinical studies and highlights the need for further understanding of receptor regulation in cancer cells [40,209,255]. Anticancer therapies that target both receptors at the same time or target molecules which are downstream the merged signaling pathway may be more effective.
Major components of the insulin signaling pathway (e.g., RAS, AKT, PI3K) are frequently mutated in cancer [41,256]. Moreover, phosphatases like PTEN which negatively regulate insulin signaling, are well-known as tumor suppressors and are also frequently mutated in cancers [41,256]. The frequent mutations in insulin signaling pathway can affect cell proliferation and survival in multiple steps. When insulin binds to INSR, it causes receptor autophosphorylation and activation of the PI3K/AKT/mTOR signaling cascade [42]. AKT can directly phosphorylate BCL2 associated agonist of cell death and caspase-9 which inhibit the mitochondrial apoptosis pathway and promote cell survival [41,256]. AKT can also indirectly inhibit apoptosis and cause cell-cycle arrest by phosphorylating and inhibiting FOXO, as FOXO can promote Bcl-2-like protein 11 and induce proapoptotic cytokine Fas ligand expression [256]. MDM2 proto-oncogene is also phosphorylated by AKT and inhibits the tumor suppressor TP53 [257]. Therefore, insulin can provide survival signals to cancer cells to escape cell-cycle arrest and apoptosis. In addition, insulin also supports cancer cell proliferation. When mTOR is phosphorylated by AKT, it activates substrates S6 kinase 1 and eIF4E-binding protein 1, which regulate mRNA translation initiation and progression to control protein synthesis and cell growth [42]. Moreover, tumor cells generally need to take up more glucose to generate biomass and support their repaid growth, so hyperinsulinemia may also contribute to cancer development through making more glucose transporters available in tumor cell surface to transport glucose. In addition to PI3K/AKT/mTOR signaling pathway, hyperinsulinemia can also promote cancer development through MAPK/ERK pathway which is important for cell proliferation and RAS is frequently mutated to be constitutively active in cancer cells [41,43,211,258]. RAS needs to be farnesylated and anchored at plasma membrane before it activated by SOS, which is a guanine nucleotide exchange factor [259]. Insulin can phosphorylate and active farnesyltransferase which is the enzyme involved in isoprenylation of RAS [259-263], so hyperinsulinemia may augment the amount of farnesylated RAS available for GTP loading in response to stimulation by other growth factors. As a result, hyperinsulinemia may promote cancer cell growth and prevent cancer cell death through both PI3K/AKT/mTORC and MAPK/ERK signaling cascades.
HYPERINSULINEMIA AND PANCREATIC CANCER
Pancreatic cancer, more specifically exocrine pancreatic cancer or pancreatic ductal adenocarcinoma (PDAC), is the 4th most common cause of cancer death [264]. Risk factors for pancreatic cancer include obesity, T2DM, pancreatitis, smoking, and family history [265-271]. As obesity and diabetes rates skyrocket, these are becoming even more ominous risk factors [265], with obesity predicted to overtake smoking as the leading preventable cause of cancer [272]. T2DM was shown to be an independent risk factor for pancreatic cancer in many studies [273-278] and some authors have implicated hyperglycemia in cancer pathogenesis [279,280]. However, inferring causality for this observation is further complicated by the fact that PDAC itself and PDAC treatment regimens can cause diabetes [281-283]. Nevertheless, a Mendelian randomization study pointed to causal roles for BMI and fasting insulin, but not T2DM or dyslipidemia, in PDAC [284].
The pancreas is anatomically unique due to its close proximity to the source of circulating insulin. After insulin is secreted from β-cells, it is first transported through the portal circulation to the liver, where over 50% of insulin is absorbed [76]. As a result, the local pancreatic insulin concentration is approximately 10 times higher than what is found in the post-hepaticcirculation [69,70]. Insulin has potent mitogenic and antiapoptotic actions on primary and transformed cells from the endocrine pancreas, from both humans and rodents [20,181,285,286]. Previous studies have shown that there were more acinar cell mitoses around islet cells compared to other pancreatic cell types, and T1DM patients had smaller number of acinar cells [287-289]. Pre-clinical islet transplant experiments have suggested that local hyperinsulinemia alone, at levels that do not cause hypoglycemia, can promote neoplasia [290,291]. Clinical and epidemiological studies showed hyperinsulinemia was associated with increased risk of cancer, including PDAC. For example, in a 16 years follow-up study for male Finnish smokers, researchers found insulin concentration in the highest vs lowest quartile predicted a 2-fold increased risk for pancreatic cancer and the associations were stronger when the follow-up was longer than 10 years [219]. Retrospective studies show that metformin, which reduces hyperinsulinemia, may lower pancreatic cancer risk in animals [292] and in patients by up to 60% [182,293-297], although the magnitude of this effect is controversial [298,299] and it is unclear whether metformin acts via lowering insulin or modulating AMP-activated protein kinase (AMPK) or another mechanism [300-303]. Thus, multiple groups have proposed that excess insulin levels contribute to pancreatic cancer initiation or progression [304-308], but this possibility had never been formally tested before our recent work [31], in part because animal models with precise control of insulin secretion or action were not available.
In vivo animal studies are required to directly test for causality in the hyperinsulinemia-cancer hypothesis. In our recent study, we demonstrated hyperinsulinemia contributes causally to pancreatic cancer development using a unique mouse model [31]. We again used female mice with lower fasting insulin without affecting the glucose homeostasis (Ins1+/−; Ins2−/− compared to Ins1+/+; Ins2−/−) [20,30] that also expressed commonly used pancreatic cancer susceptibility alleles that reflect human disease genetics (Ptf1aCreER; LSL-KrasG12D) [258]. We fed all groups of mice a hyperinsulinemia-inducing high-fat diet. We found there were significant reductions of pre-pancreatic cancer lesions and fibrogenesis in mice with reduced insulin secretion, demonstrating that endogenous hyperinsulinemia contributes to pancreatic cancer development [31]. These observations are consistent with other, less direct models designed to test whether hyperinsulinemia accelerates the initiation and/or progression of other types of cancer. The next step will be to determine which of the main Insr expressing cell types contribute to the effects of hyperinsulinemia on pancreatic cancer initiation and/or progression (Fig. 2B).
TESTING THE INSULIN-CANCER HYPOTHESIS OUTSIDE OF THE PANCREAS
Insulin signaling connects with many signaling networks known to drive breast cancer initiation and progression (Fig. 3). Epidemiological evidence demonstrates that both obesity and diabetes are associated with breast cancer incidence and prognosis. Clinically, breast cancer is categorized into three subtypes, which dictate treatment courses and prognoses. More than 70% of breast cancers (>150,000 cases/year) overexpress the estrogen receptor (ER) and/or progesterone receptor (PR) and these are frequently diagnosed after menopause [309]. Half of the remaining tumors express the human epidermal growth factor receptor 2 (HER2). The other half of cases lack all three receptors and are classified as triple negative [309]. The obesity-specific risk for breast cancer is greatest for the ER/PR-positive subtype [310]. Breast cancer risk increases by 12% (relative risk [RR], 1.12) for every 5 kg/m2 increase in BMI [269]. Because obesity is a shared risk factor between diabetes and breast cancer, most studies evaluate cancer risk in patients with diabetes after adjusting for BMI. Data from the Nurses’ Health Study suggest that women with diabetes have an elevated risk for breast cancer (hazard ratio [HR], 1.17) [311], which was confirmed in a more recent meta-analysis (summary relative risk [SRR], 1.16) [312]. These significant relationships remained after adjusting for BMI. A different study found that diabetes was associated with increased breast cancer-specific mortality (RR, 1.27) [312,313] that was not explained by obesity. Both obesity and diabetes are associated with advanced breast cancer at diagnosis [310, 314,315], including larger, higher grade tumors and lymph node involvement [310,314,316]. In each study that found a relationship between breast cancer and either obesity or diabetes, the link was strongest in postmenopausal women [269,312, 316-319]. Before menopause, obesity and diabetes each associate with a decreased risk for breast cancer [312,317], but the mechanisms behind this paradox are unclear. Importantly, the associations between diabetes and breast cancer are specific to T2DM, as individuals with T1DM do not face increased risk or mortality [312]. This observation implies that one or more of the major pathophysiological differences between T2DM and T1DM, such as hyperinsulinemia or hyperlipidemia (but not hyperglycemia) play causal roles.
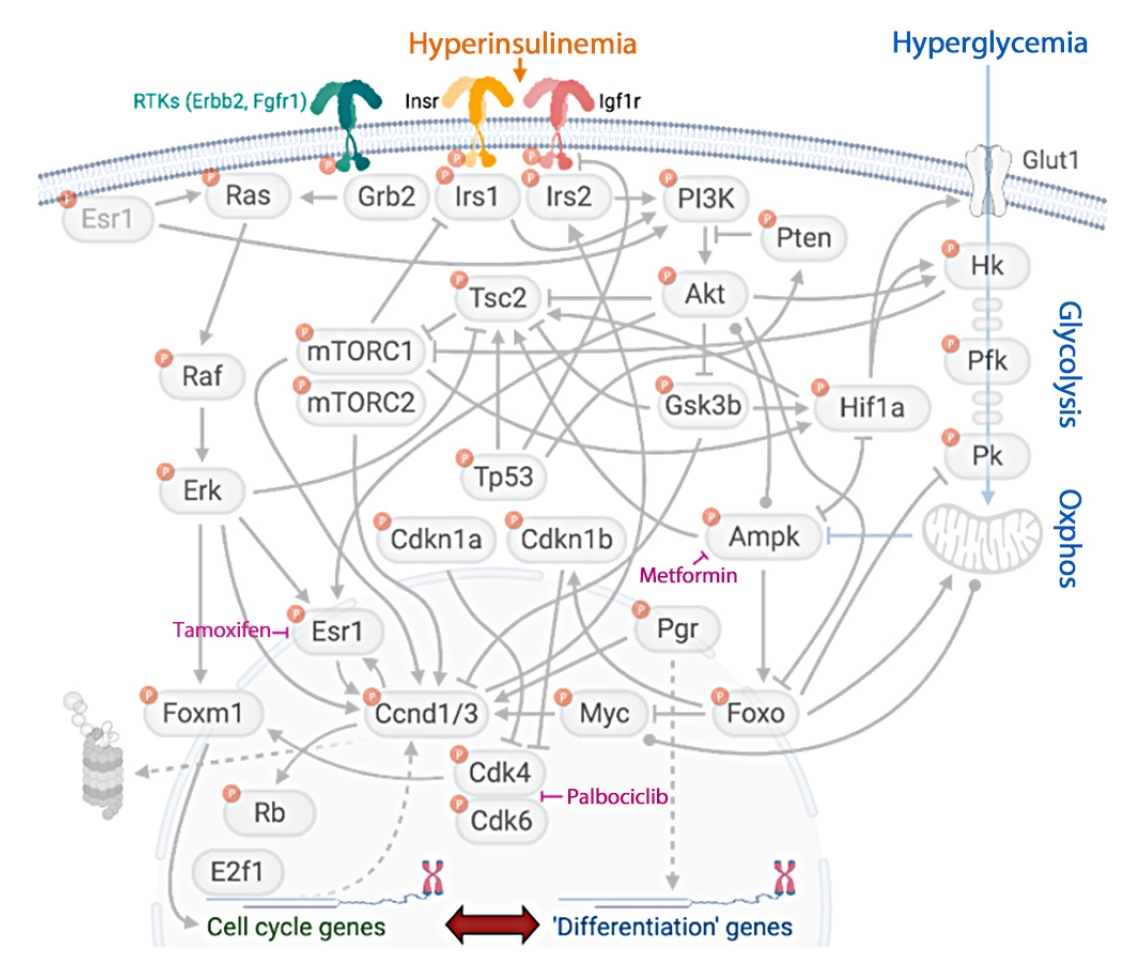
Multiple links between insulin signaling and breast cancer. Insulin/Igf1 receptor signaling interacts with master regulators of breast cancer cell fate, including estrogen receptor signaling, core cell-cycle regulators, and metabolism. Virtually all proteins in this signaling network are regulated by phosphorylation. RTK, receptor tyrosine kinase; Erbb2, proto-oncogene Neu/Her2; Fgfr, fibroblast growth factor receptor; Insr, insulin receptor; Esr1, estrogen receptor 1; Grb2, growth factor receptor-bound protein 2; Irs, insulin receptor substrate; PI3K, phosphoinositide 3-kinase; Tsc2, tuberous sclerosis complex 2; Pten, phosphatase and tensin homolog; Glut1, glucose transporter 1; Hk, hexokinase; mTORC, mammalian target of rapamycin complex; Erk, extracellular signal-regulated kinase; Tp53, tumor protein p53; Gsk3b, glycogen synthase kinase 3b; Hif1a, hypoxia-inducible factor 1a; Pfk, phosphofructokinase; Pk, pyruvate kinase; Cdkn1, cyclin-dependent kinase inhibitor-1; Ampk, AMP-activated protein kinase; Pgr, progesterone receptor; Foxm1, forkhead box M1; Ccnd1/3, cyclin-dependent kinase 1/3; Myc, myc oncogene; Foxo, forkhead family box O; Cdk, cyclin-dependent kinase; Rb, retinoblastoma; E2f1, E2F transcription factor 1.
Two underlying drivers of the obesity-cancer relationship may be metabolic dysfunction and adult weight gain. Three studies found similar links between metabolic health and breast cancer risk independent of obesity. In a prospective cohort study, women with a normal weight (BMI <25 kg/m2) and one or more features of metabolic disease (e.g., high waist circumference, elevated blood pressure, and/or diabetes) had a similarly elevated risk for postmenopausal breast cancer (HR, 1.26) to women who were metabolically healthy but had overweight or obesity (HR, 1.24) [320]. Another recent study found that obesity, regardless of metabolic health, associated with increased risk of breast cancer, with the highest risk seen in metabolically unhealthy women with obesity (HR, 1.62) [321]. A third study found that women classified as metabolically unhealthy based on homeostatic model assessment for insulin resistance or fasting insulin had an elevated risk for breast cancer regardless of BMI [197]. The most recent, large, prospective cohort study reported elevated risks for breast (HR, 1.16) and endometrial cancer (HR, 2.94) in women with overweight or obesity even after adjusting for metabolic status [322]; however, risks were greatest in the presence of metabolic disease and obesity [315]. Together, these studies suggest that while metabolic dysfunction contributes to cancer, it may not be the only mechanism. Women who transitioned from a “lean” BMI (<25 kg/m2) to either overweight or obese faced an increased risk of breast cancer (HR, 1.36) compared with women who had an elevated BMI as young adults and did not gain more than 5% weight over approximately 13 years of follow-up [316]. In addition, a meta-analysis estimated that every 5 kg of weight gained increased the risk for breast cancer by 11% (SRR, 1.11) and endometrial cancer by 39% (SRR, 1.39) [323].
As with obesity, women with diabetes may have a greater risk of breast cancer recurrence compared to women without diabetes (odds ratio [OR], 2.21) [314]. However, whether diabetes increases breast cancer-specific mortality remains controversial. A recent Mendelian randomization study that investigated the genetic predisposition to T2DM, as well as predicted fasting insulin and glucose, did not find a causal association with any variable and breast cancer risk [324], but this does not preclude roles for non-genetic drivers of hyperinsulinemia and hyperglycemia. Some studies [325] including the Women’s Health Initiative (WHI) [315,326], do not support a link between diabetes and death specifically from breast cancer. In contrast, one large prospective study found that diabetes increased the risk for death from breast cancer in women (RR, 1.16) and in men (RR, 4.20) [200] after adjusting for BMI. Similarly, a smaller population-based study found that diabetes predicted a shorter breast cancer specific survival (HR, 1.53) for women with early stage breast cancer, which was also independent of BMI [327]. Consistent with breast cancer, obesity and diabetes are strongly associated with the incidence and mortality of endometrial cancer [328-330]. Obesity is thought to be responsible for over half of endometrial cancer diagnoses [269,331], with risk increasing by 50% (RR, 1.50) for every 5 kg/m2 increase in BMI [332]. Data from the WHI suggests that elevated BMI increases the risk for endometrial cancer by 76% (HR, 1.76) [333]. Diabetes increases the risk for endometrial cancer by 2-fold [328], and a Mendelian randomization analysis estimated a small increase (OR, 1.08) in the risk associated with both diabetes and predicted fasting insulin [324]. However, among a total of nine studies examining diabetes and endometrial cancer risk, only four studies [328,334-336] adjusted for BMI with two [328,336] reporting a significant association independent of BMI. Thus, for endometrial cancer, the data are moderate that diabetes is associated with risk independently of obesity. Unlike breast cancer, T1DM is associated with endometrial cancer risk (SRR, 3.15) [328], suggesting subtle but potentially important underlying differences in etiology.
Elevated fasting insulin and glucose are commonly associated with diabetes, obesity, and weight gain. Consistent with epidemiological observations, insulin and glucose have also been attributed to breast and endometrial cancer. Genetically predicted fasting insulin levels were associated with endometrial cancer risk after adjusting for BMI in a Mendelian randomization study [324]. Hyperinsulinemia associated with an increased risk for breast (HR, 1.46) [337] and endometrial cancer (HR, 2.33) [228] in the WHI study. For endometrial cancer, the risk was higher in women with hyperinsulinemia and overweight or obesity (HR, 4.30) [228]. For both cancer types, this relationship between insulin and risk was specific to nonusers of menopausal hormone replacement therapy [228,337], suggesting potential crosstalk between the insulin and estrogen signaling pathways in driving early tumor growth. A different study reported that insulin resistance, inferred from C-peptide levels, associated with elevated breast cancer risk in women without diabetes (RR, 2.9) [338]. The potential tumor promoting mechanisms downstream of insulin are similar between breast and endometrial cancers, and include extensive cross talk with signaling networks regulated by E2 and IGF1 [338,339]. Insulin itself is a potent mitogen for cancer cells [340,341], and may potentiate signaling through ER [338,340,342]. Data from large randomized controlled trials suggest that improving glycemic control does not reduce the risk for breast cancer [343], although this has been debated [344]. The large body of evidence linking obesity and diabetes to women’s cancers has supported clinical trials that aim to address the efficacy of metformin, a widely used anti-hyperglycemic agent, against breast [345-347] and endometrial cancers [348].
Hyperinsulinemia-induced by muscle cell-specific insulin resistance in mice [349] was found to accelerate esophageal cancer [38] and breast cancer development and increase lung metastases [5,45-47,209,350]. The tissues and cancer cells from these hyperinsulinemic mice showed increased activation of INSR/PI3K/AKT signaling pathway and mammary tumor growth was independent of the IGF1 receptor [47,351]. Exogenous insulin promoted breast and colon carcinogenesis [352,353], and diet-induced hyperinsulinemia was able to accelerate prostate cancer xenograft progression [354]. Additionally, endometrial carcinoma cell growth could be promoted by overexpressing INSR, and pancreatic cancer cells proliferated proportionally with the increase of insulin concentrations [215,240,355]. Knocking out INSRs slowed the growth of pancreatic neuroendocrine cancer cells and melanoma cells as well as inducing the apoptosis in DNA-damaged colon cells [356-358]. Therefore, in addition to epidemiological data, the experimental mouse models provide abundant evidence that hyperinsulinemia can contribute to carcinogenesis. It remains unclear whether these effects of insulin are via cancer initiating cells, local immune cells, cancer associated stroma, or indirectly through insulin’s effects on adiposity. Likely, multiple complex mechanisms are responsible for specific cancer promotion in various tissue types. Experiments where INSR is deleted from these putative cellular mediators will be required to provide clarity to this question.
Hyperinsulinemia may also contribute to cancer development through influencing IGF1 levels. IGF1, which plays a critical role in the development of various tumors, is primarily produced in liver under the stimulation of growth hormone [212,359-362]. Similarly, insulin can also increase sex hormone and reduce sex hormone binding protein production [211,363]. This increase of hormones such as estrogen and androgen can affect the growth of hormone-dependent tumors in the breast, prostate, and endometrium. As mentioned above, hyperinsulinemia is correlated with inflammation, which can contribute to tumorigenesis in many different aspects [364,365]. Together, the studies discussed above provide potential mechanisms by which hyperinsulinemia may contribute to cancer development.
As people become more aware of the cause-and-effect relationship between insulin and cancer, many more anticancer therapies targeting hyperinsulinemia have been developed and tested in animal models or in clinical trials. For instance, a recent study showed that the combination of PI3K inhibitors with kenogenetic diet or sodium-glucose transport protein 2 (SGLT2) inhibitors can effectively reduce tumor growth and increase the survival rate [366]. Also, multiple studies have demonstrated metformin could possess anti-cancer properties and there are some drugs in pre-clinical trials already [367,368]. In summary, although it is still at an early stage, our studies have demonstrated that hyperinsulinemia can contribute to tumorigenesis. It follows that lifestyle interventions or therapeutics with mild insulin suppressing actions may be useful in the prevention and treatment of some types of cancers.
CONCLUSIONS
Hyperinsulinemia is a condition associated with obesity and early stage T2DM. Recent studies have implicated hyperinsulinemia in multiple pathological conditions including insulin resistance, inflammation, obesity, and cancer. Modest inhibition of insulin production or insulin signaling is sufficient to increase of lifespans in a variety of animal models, from invertebrates to mice. Additional studies are required to elucidate the relationship between insulin and IGF during aging, as well as the shared and distinct molecular mechanisms. Conventionally, hyperinsulinemia was considered to be an adaptation to obesity-induced insulin resistance. However, evidence continues to mount that hyperinsulinemia can precede and cause obesity and insulin resistance. Animal models with reduced endogenous insulin secretions were protected from diet-, age-, and leptin-induced obesity, which clearly demonstrates the causal role of hyperinsulinemia in obesity. It is also clear that insulin resistance can lead to hyperinsulinemia, including at the level of the pancreatic β-cells, suggesting at least a bi- or tri-directional relationship. Future studies will be required to determine the nature of the complex vicious cycles that lead to T2DM. The metabolic importance of insulin has been long recognized but now the field is beginning to appreciate its importance in cancers. Both clinical and epidemiological studies demonstrated hyperinsulinemia is associated with increased cancer morbidity and mortality. Furthermore, direct animal studies have shown hyperinsulinemia could promote tumorigenesis, especially for pancreatic. Invertebrate models of hyperinsulinemia-induced cancer provide opportunities for powerful screens [369] that may uncover the specific molecular mechanisms involved and lead to targeted therapeutics. In summary, while insulin is essential for maintain normal life, the negative consequences of hyperinsulinemia shed light on the importance of maintaining insulin levels within a healthy range. Lifestyle interventions or therapeutics with mild insulin suppressing actions provide new opportunities to prevent and treat certain disorders like obesity, chronic inflammation and cancers.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
None
Acknowledgements
None