- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 37(6); 2013 > Article
-
ReviewObesity and Metabolic Syndrome Targeting the Peroxisome Proliferator-Activated Receptor-γ to Counter the Inflammatory Milieu in Obesity
- Cesar Corzo, Patrick R. Griffin
-
Diabetes & Metabolism Journal 2013;37(6):395-403.
DOI: https://doi.org/10.4093/dmj.2013.37.6.395
Published online: December 12, 2013
Department of Molecular Therapeutics, The Scripps Research Institute, Jupiter, FL, USA.
- Corresponding author: Patrick R. Griffin. Department of Molecular Therapeutics, The Scripps Research Institute, Scripps Florida, 130 Scripps Way #2A2, Jupiter, FL 33458, USA. pgriffin@scripps.edu
Copyright © 2013 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Adipose tissue, which was once viewed as a simple organ for storage of triglycerides, is now considered an important endocrine organ. Abnormal adipose tissue mass is associated with defects in endocrine and metabolic functions which are the underlying causes of the metabolic syndrome. Many adipokines, hormones secreted by adipose tissue, regulate cells from the immune system. Interestingly, most of these adipokines are proinflammatory mediators, which increase dramatically in the obese state and are believed to be involved in the pathogenesis of insulin resistance. Drugs that target peroxisome proliferator-activated receptor-γ have been shown to possess anti-inflammatory effects in animal models of diabetes. These findings, and the link between inflammation and the metabolic syndrome, will be reviewed here.
- The percentage of the global population that is obese has skyrocketed over the past few decades and it is predicted that this percentage will continue to rise significantly as the developing world adopts more sedentary lifestyles and gains access to high calorie diets. As recently as 10 years ago in the United States, obese adults (defined as body mass index >30) made up less than 12% of the population. Now, more than 20% of the adult population meets the Centers for Disease Control and Prevention criteria for obesity and greater than 40% are considered overweight. The metabolic syndrome is characterized as a clustering of factors associated with an increased risk of cardiovascular disease and stroke and is becoming more common [1]. The essential risk factors that constitute the metabolic syndrome are age, atherogenic dyslipidemia (high triglycerides and low high density lipoprotein cholesterol), hypertension, elevated plasma glucose, a prothrombotic state, and a proinflammatory state. The two major underlying causes of the metabolic syndrome are obesity and type 2 diabetes mellitus (T2DM) [2]. Obesity is conceptually defined as an excess of body lipids of sufficient magnitude to impair health or longevity. T2DM is a chronic metabolic disorder that is caused, in part, by the inability of the body to respond adequately to circulating insulin, a condition termed insulin resistance. The comorbidities of the metabolic syndrome will continue to strain global health care systems requiring development of strategies to combat this epidemic.
- The therapeutic approach for the treatment of the metabolic syndrome is to identify and treat each risk factor separately. Current standard of care includes pharmacologic intervention and lifestyle modification such as nutritional consultation and modification of diet, rigorous weight control, and increased regular exercise. Modest weight loss can result in an improvement in metabolic parameters [3]. However, weight loss and exercise often are not sufficient due to poor compliance or perhaps genetic factors [4,5]. Pharmacologic approaches have focused on the use of combination therapy for diabetes treatment in an effort to postpone the inevitable need for insulin therapy [6]. Molecular targets for treatment of the metabolic syndrome include the PPAR subfamily of nuclear receptors. Here we review current thoughts on the role of the γ isoform of the PPAR subfamily in metabolic disease. We also review recent mechanistic studies that pave the pathway to develop novel approaches to pharmacologically target PPAR-γ for the treatment of the metabolic syndrome.
INTRODUCTION
- Nuclear receptors (NRs) are a major target of drug discovery. NRs are ligand-dependent transcription factors that possess the ability to directly interact with DNA and they regulate DNA transcription, in part, by recruitment of chromatin remodeling enzymes to promoters of target genes. These receptors play essential roles in development, cellular homeostasis and metabolism [7]. Moreover, NRs have been implicated in a wide range of diseases and, as such, have been the focus of drug development efforts for the pharmaceutical industry. One of the NR families intimately involved in maintaining metabolic homeostasis is the peroxisome proliferator-activated receptor (PPARs) or NR1C subfamily. The PPAR subfamily consists of three members encoded by different genes and is involved in a myriad of physiological processes that impact lipid homeostasis, inflammatory responses, adipogenesis, insulin sensitivity, reproduction, wound healing, and carcinogenesis [8,9].
- PPAR-α was first characterized as mediating the effect of peroxisome proliferators [10]. Subsequently, the other closely related receptors were cloned and named PPAR-γ and PPAR-δ despite neither responding to peroxisome proliferators [11]. Similar to other NRs, PPARs possess a N-terminal ligand-independent transactivation domain (AF-1), a two zinc-finger DNA-binding domain, a hinge domain, and a C-terminal ligand-binding domain containing a ligand responsive activation domain (AF-2). The PPARs have been shown to heterodimerize with the 9-cis-retinoic acid receptor α [12] and regulate transcriptional activity by binding to direct repeat 1 PPAR response elements (5' AGGTCA (N)X AGGTCA 3') in the promoter or enhancer regions of its target genes [13]. The PPAR family members are differentially expressed with PPAR-α being highly abundant in tissues with high potential for fatty acid oxidation such as the skeletal muscle, liver, kidney, heart, and brown adipose tissue. It regulates peroxisome proliferation, lipid catabolism, inflammatory responses, and skin wound healing. PPAR-δ is ubiquitously expressed and is suggested to influence myelination, embryonic implantation, and adipocyte differentiation. In addition, PPAR-δ activity contributes to the restoration of metabolic homeostasis by improving hypertriglyceridemia and insulin resistance, controlling body weight, and suppressing inflammation [14]. PPAR-γ is mainly expressed in adipose tissue where it plays a critical role in adipocyte differentiation and lipogenesis. PPAR-γ is also expressed in monocytic cells and its modulation by synthetic ligands can influence inflammatory processes. We will briefly review the importance of PPAR-γ in inflammation and its connection to the metabolic syndrome observed in obesity.
PPARs AND METABOLIC DISORDERS
- One of the main contributing factors to the development of the metabolic syndrome in diabetics is the excessive expansion and enlargement of adipocytes. This results in the increased secretion of inflammatory factors that can interfere with insulin signaling and glucose disposal. Insulin secreted from pancreatic islet β-cells interacts with its receptor in its major target tissues which are skeletal muscle, liver, and adipose tissue [15]. Insulin is arguably the most important hormone influencing lipogenesis. In order to maximize glucose uptake and clearance, insulin must regulate the availability of free fatty acids in circulation. It does so by opposing the rate of lipolysis in adipose tissue and stimulating the re-esterification of fatty acids into triglycerides for storage. The expression of enzymes involved in fatty acid and triglyceride synthesis, such as acetyl-CoA carboxylase, fatty acid synthase, and diacylglycerol acyltransferase, are induced upon activation of the insulin receptor (IR) [16].
- The IR is a heterotetramer consisting of two extracellular α-subunits and two transmembrane β-subunits held together by disulfide bonds [17]. The intracellular portion of the protein contains a tyrosine kinase domain and phosphotyrosine binding sites for signaling molecules. Insulin binding to the extracellular portion of the receptor induces conformational changes in the protein which activate the tyrosine kinase domain in the intracellular region. Activation of the kinase domain results in rapid phosphorylation of intracellular insulin receptor substrates (IRS 1 to 4) that serve as docking sites for downstream signaling molecules [18]. The metabolic effects of insulin are mediated through the PI3k-Akt-PKC pathway which is critical for proper translocation of the insulin-regulated glucose transporter GLUT4 to the plasma membrane [19]. Whereas tyrosine phosphorylation of the docking proteins is indispensable for efficient insulin action, serine/threonine phosphorylation of IRS1 blocks proper downstream signaling and contributes to the desensitization to insulin in T2DM [20]. The chronic low-grade inflammatory state associated with obesity is known to interfere with IR signaling, contributing to the development of insulin resistance. Development of this inflammatory state is initiated, in part, by the release of proinflammatory mediators released from adipocytes that have become excessively large. Stress and inflammatory pathways are subsequently activated by these mediators resulting in the infiltration of a heterogeneous population of cells into adipose tissue consisting of lymphocytes, granulocytes, and monocytes.
- Monocytes develop in the bone marrow where they differentiate from common myeloid progenitors. After differentiation, monocytes are released into circulation where they rapidly move to the site of inflammation upon receiving proper migratory signals. The chemokine receptor CCR2, a prominent receptor for the monocyte chemoattractant protein 1 (MCP-1) plays a key role in the migration and extravasation of monocytes. In response to insulin, 3T3-L1 mouse preadipocyte fibroblasts secrete substantial amounts of MCP-1. Injection of insulin into rodents also results in a substantial increase in circulating MCP-1 [21]. Relative to liver, kidney, and lung, fat from obese leptin-deficient ob/ob mice expresses high levels of MCP-1 indicating that adipose tissue is a major producer of this chemokine. Thus, MCP-1 is likely a major factor involved in the recruitment of monocytic cells into adipose tissues in the obese setting. In fact, the percentage of the monocyte population in adipose tissue can reach up to 50% in the ob/ob mouse and up to 40% in obese humans [22].
- Once monocytes infiltrate adipose tissue, they undergo a process of maturation into macrophages that consists of cell enlargement and development of higher mitochondrial numbers and larger lysosomal structures with increased content of hydrolytic enzymes [23]. Macrophages are found in a multitude of tissues throughout the body and are essential for normal tissue development and function [24,25]. Macrophages are also essential for metabolic homeostasis. Pancreatic macrophages help maintain appropriate islet cell morphology and proper insulin production. In adipose tissue, resident macrophages produce interleukin (IL)-10, which facilitates insulin action in adipocytes [26]. In order to maintain metabolic homeostasis, the proper function of macrophages is essential.
- Macrophages show remarkable plasticity and functional heterogeneity, as they participate in both the initiation and resolution of inflammation. This ability to perform opposite functions is controlled by the type of stimuli encountered. In the presence of type I interferon and microbial triggers, macrophages undergo classical activation, in which they are distinguishable by the generation of reactive oxygen species (ROS), nitric oxide (NO), and proinflammatory cytokines like tumor necrosis factor (TNF)-α, IL-6, and IL-12. In contrast, the type II cytokines IL-4 and IL-12 promote maturation of alternatively activated macrophages, a distinct program associated with reduced ROS generation, a diversion of arginine metabolism through upregulation of arginase I, production of the anti-inflammatory cytokine IL-10, and expression of distinct phagocytic receptors [27]. Each macrophage subtype has independent functions: the M1 subtype plays a pivotal role in the clearance of invading pathogens, whereas M2 macrophages are involved in tissue repair.
- In the lean state, resident macrophages in adipose tissue exhibit M2-like characteristics. However, as adipocytes undergo hypertrophy leading to cellular stress and apoptosis, fresh monocytes are recruited to remove the cellular debris and eventually become macrophages that form crown-like structures around the dead adipocytes. Saturated fatty acids released from adipocytes are capable of binding and inducing the toll-like receptor 4 signaling cascade [28] that ultimately results in the activation of the M1 inflammatory program and secretion of the proinflammatory cytokines TNF-α and IL-6, and the generation of NO. These activated macrophages potentiate the inflammatory response and, in turn, attract more leukocytes, forming a positive feedback loop and a state of chronic inflammation. Demonstrating the importance of macrophages in insulin sensitization and metabolic syndrome, CCR2 deficient mice show resistance to diet-induced obesity and improved glucose homeostasis. Therapeutic treatment with a selective CCR2 antagonist mimicked these effects in mice maintained on a high-fat diet [29].
- Inflammatory mediators have been extensively studied for their ability to interfere with insulin signaling in adipocytes and muscle cells, negatively altering downstream kinase-dependent signaling and GLUT4 translocation [30,31] and opposing the action of insulin in order to limit nutrient storage. Secretion of TNF-α was the initial factor proposed to link insulin resistance and inflammatory signaling. TNF-α is overexpressed in both muscle and adipose tissues from obese subjects [32,33] and appears to mediate its anti-insulinemic effects by targeting the phosphorylation status of the IR. In obese TNF-α null mice, the IR shows higher levels of tyrosine phosphorylation [34] and these mice respond more efficiently to exogenous glucose and insulin administration. Like TNF-α, IL-6 also inhibits insulin signaling in adipocytes [35]. Furthermore, inhibitory effects of IL-6 on insulin signaling have also been shown in primary hepatocytes [36].
- The role of NO in insulin resistance is more complex. NO is synthesized by the nitric oxide synthase (NOS) family of enzymes, of which there are three isoforms differentially expressed in various tissues [37]. The inducible isoform of NOS (iNOS) is activated by inflammatory mediators and there is a wealth of data supporting the notion that overproduction of NO inhibits the action of insulin. In vivo application of the NOS inhibitor L-NAME improved insulin sensitivity in mice fed a high fat diet (HFD) and also reduced the size of adipocytes accompanied with reduced triglyceride accumulation [38]. Furthermore, iNOS deficiency protected mice from HFD-induced insulin resistance and improved brown adipose tissue function. iNOS-/- obese mice displayed normal insulin sensitivity and glucose disposal [39] and iNOS deletion in ob/ob mice enhanced the expression of mitochondrial proteins such as uncoupling proteins UCP1 and UCP3, and the transcription factor PRDM16 [40], reducing body weight and weight of total fat pads, and increasing rectal temperatures.
- While PPAR-γ positively regulates the anabolism of lipids in macrophages, activation of PPAR-γ acts as a negative regulator of the inflammatory mediators mentioned above. This inhibitory function appears to be mediated by sumoylation of PPAR-γ at K365 [41]. Pascual et al. [41] demonstrated that sumoylation at K365 enhances the interaction of PPAR-γ with the nuclear corepressor (NCoR). Certain promoters, such as those controlling inflammatory genes, were shown to have NCoR-HDAC3-TBL corepressor complexes present in the basal state. Stimulation with mediators such as lipopolysaccharide triggered removal of these repressor complexes from DNA allowing the initiation of transcription of inflammatory genes. Increased sumoylation of PPAR-γ resulted in retention of repressor complex at these promoters and a reduction in transcription of inflammatory genes.
INFLAMMATION IN ADIPOSE TISSUE
- As mentioned above, macrophages are highly adaptable due to their extreme plasticity, which allows them to switch between M1 and M2 phenotypes upon stimulation. Hence, it is tempting to speculate that manipulation of this attribute of macrophages could ameliorate the inflammatory milieu in adipose and muscle tissue of obese individuals and restore normal insulin sensitivity. PPAR-γ could provide such a molecular switch necessary to return macrophage polarization back to the M2 state. PPAR-γ is expressed in macrophages and its genetic disruption in murine monocytes predisposes mice to insulin resistance and glucose intolerance [42]. Additionally, PPAR-γ null macrophages have a reduced rate of fatty acid oxidation and reduced number of mitochondria. PPAR-γ, therefore, is essential to macrophages for the molecular transition to alternative polarization.
- The thiazolidinediones (TZDs) are potent PPAR-γ activators (potent full agonists) that are used clinically for the treatment of T2DM. TZDs increase the expression of many proteins within the insulin signaling cascade such as the IR docking proteins, PI3-kinase, glucose transporters 1, and 4. TZD treatment also leads to a reduction of circulatory levels of low density lipoprotein and triglycerides [43-45]. An additional property of TZDs is their anti-inflammatory effects in adipose tissue where TZDs attenuate production of several inflammatory mediators including TNF-α, IL-6, and iNOS [46]. Recently Nguyen et al. [47] showed that a switch to the M2 phenotype in brown and white adipose tissue improves the adaptation to cold temperatures as IL-4-stimulated macrophages release noradrenaline, which facilitates fatty acid mobilization and energy expenditure.
POLARIZATION OF MACROPHAGES AS A STRATEGY FOR IMPROVING OF METABOLIC DISORDERS
- Unfortunately, the beneficial effects of TZDs in terms of improvement of glucose homeostasis, insulin sensitivity, and lipid profile is largely negated by well documented side effects such as fluid retention which is associated with increase for heart failure and the propensity for bone fractures. The increased incidence of bone fractures in patients on TZD treatment likely results from the ability of PPAR-γ to act as a negative regulator of osteoblastogenesis; hence, its hyper activation (superaphysicalogical agonism) carries negative skeletal effects. In cultured murine and human cells, the activation of PPAR-γ and its overexpression induces bone marrow stromal cell differentiation into the adipocyte lineage [48]. The use of the PPAR-γ agonist pioglitazone in ovariectomized rats reduced whole body and femoral bone mineral density [49]. Clinical studies have also provided evidence of the negative effect of TZDs on bone formation [50,51].
- Systemic fluid retention is a major factor that has complicated the clinical use of TZDs. PPAR-γ is highly abundant in the collecting duct system of the kidney. Sodium channels present in the collecting duct have been shown to be target genes of PPAR-γ and their expression increases upon stimulation with TZDs resulting in increased Na+ absorption. The reduced Na+ excretion has been accredited with increased systemic water retention or plasma volume expansion (PVE). Increased total body water is partly responsible for the early weight gain observed in animal models treated with TZDs and weight gain can be prevented by deletion of PPAR-γ in the collecting ducts. In addition to weight gain, additional complications of PVE are edema and cardiomegaly. In clinical trials the incidence of edema is greater when TZDs are used in combination with other glucose-lowering agents, such as metformin, sulfonylurea, and insulin [52,53]. In preclinical trials, cardiac hypertrophy has been observed after TZD treatment. The cardiac effects are independent of insulin signaling or PPAR-γ expression in the heart and appear to stem from the cardiac volume overload (increased back pressure) caused by PVE [54].
- Because of these side effects the use of TZDs has been restricted or, in some cases, the compounds have been withdrawn from the market altogether. This is unfortunate as this class of compounds have demonstrated clear robust efficacy. Thus, significant efforts had gone into the development of second-generation PPAR-γ activators called selective PPAR-γ modulators or SPPARγMs. In general, partial agonists of PPAR-γ demonstrate the SPPARγMs phenotype in preclinical models. In such models, SPPARγMs demonstrate a clear advantage over TZDs in terms of their side effect profile; however, clinical development of all SPPARγMs have been discontinued.
TZDs, THEIR SIDE EFFECTS, AND SPPARγMs
- Similar to other NRs, PPAR-γ activity is regulated at the posttranslational level by modifications at key residues [55,56]. Obesity, in particular, promotes the phosphorylation of PPAR-γ at S273 (pS273), and this posttranslational modification (PTM) is correlated with the dysregulation of a subset of PPAR-γ target genes, many of which are dysregulated in obesity. pS273 sensitive genes include the insulin-sensitizing adipokines such as adiponectin and adipsin [57]. It was shown that partial agonist SPPARMγS block pS273 to the same extent of full agonists TZDs and there was a strong correlation in both obese mice and obese humans between the antidiabetic effects of drug treatment and the reduction of pS273. These results suggest that efficacy may be derived from blockage of this PTM and not from activation of the receptor itself, which partially explains the improved side effect profile of the partial agonist SPPARγMs. Based on this discovery, and in collaboration with Bruce Spiegelman, we set out to develop compounds that were non-agonists (neutral antagonists) of the receptor but retained the ability to block pS273. This effort led to the development of SR1664. We demonstrated that the antagonist SR1664 was equally efficacious as TZDs in correcting elevated plasma glucose and fasting insulin levels in diabetic mice. We also demonstrated that SR1664 was antiadipogenic and did not cause weight gain or PVE in the same mice. Cell culture experiments demonstrated that SR1664 was neutral on bone whereas TZD treatment was detrimental, as expected [58,59]. While SR1664 offers hope for targeting PPAR-γ with significantly improved therapeutic index, the pharmaceutical properties of this compound warrant significant optimization. As such, we have continued the development of compounds with similar functional properties of SR1664, but with significantly improved pharmacokinetics.
- It is plausible that such neutral PPAR-γ antagonists that maintain the ability to block pS273 will retain the transcriptional repressive properties of TZDs in the macrophage, thus maintaining the ability to dampen the inflammation that accompanies obesity (Fig. 1). Such a compound would normalize expression of adipokines repressed by pS273, improve IR signaling, yet be devoid of adipogenesis and fluid retention, and will be neutral on bone density. The functional properties sought for an ideal compound are displayed in Table 1.
ALTERNATIVE APPROACHES TO MODULATING PPAR-γ
- The PPAR subfamily of nuclear receptors plays major roles in the energy homeostasis by promoting either the utilization or the storage of the energy sources glucose and free fatty acids. Several drugs targeting PPAR-γ for the treatment of T2DM have been developed and used clinically. Unfortunately, due to significant concerns over side effects, the utility of these drugs are limited. Significant effort had gone into the development of second-generation compounds but none of these have made it to the market. All of the second-generation compounds were either selective PPAR-γ agonists (partial or full agonists) or dual or pan PPAR modulators activating alpha and delta. Recent studies show that blockage of a specific PTM, and not activation of the receptor, correlates with the efficacy of PPAR-γ modulators. These studies open up the possibility that antagonists of PPAR-γ may offer the same potential to improve insulin sensitivity without activation of pathways responsible for the many side effects of PPAR-γ agonists, such as weight gain, water retention, and bone loss. One such compound, SR1664, that blocks phosphorylation of S273 and is antidiabetic yet does not activate PPAR-γ target genes, has been described. Efforts continue to optimize this chemical scaffold for both potency and pharmaceutical properties. We are hopeful that these efforts will lead to a new generation of safer antidiabetic drugs.
CONCLUSIONS
- 1. Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med 2005;56:45-62. ArticlePubMed
- 2. Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC Jr, Spertus JA, Costa F. American Heart Association. National Heart, Lung, and Blood Institute. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005;112:2735-2752. ArticlePubMed
- 3. Grundy SM, Hansen B, Smith SC Jr, Cleeman JI, Kahn RA. American Heart Association. National Heart, Lung, and Blood Institute. American Diabetes Association. Clinical management of metabolic syndrome: report of the American Heart Association/National Heart, Lung, and Blood Institute/American Diabetes Association conference on scientific issues related to management. Circulation 2004;109:551-556. ArticlePubMed
- 4. Moller DE, Bjorbaek C, Vidal-Puig A. Candidate genes for insulin resistance. Diabetes Care 1996;19:396-400. ArticlePubMedPDF
- 5. Bouchard C. Genetic factors in the regulation of adipose tissue distribution. Acta Med Scand Suppl 1988;723:135-141. ArticlePubMed
- 6. Gavin JR 3rd. How can we implement current therapies and interventions to achieve glycemic control? Endocr Pract 2006;12(Suppl 1):93-97. ArticlePubMed
- 7. Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 2004;3:950-964. ArticlePubMedPDF
- 8. Escher P, Wahli W. Peroxisome proliferator-activated receptors: insight into multiple cellular functions. Mutat Res 2000;448:121-138. ArticlePubMed
- 9. Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci 2004;61:393-416. PubMed
- 10. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990;347:645-650. ArticlePubMedPDF
- 11. Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992;68:879-887. ArticlePubMed
- 12. Chan LS, Wells RA. Cross-talk between PPARs and the partners of RXR: a molecular perspective. PPAR Res 2009;2009:925309ArticlePubMedPMCPDF
- 13. Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992;358:771-774. ArticlePubMedPMCPDF
- 14. Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest 2006;116:590-597. PubMedPMC
- 15. Kaplan SA. The insulin receptor. J Pediatr 1984;104:327-336. ArticlePubMed
- 16. Wong RH, Sul HS. Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr Opin Pharmacol 2010;10:684-691. ArticlePubMedPMC
- 17. Lawrence MC, McKern NM, Ward CW. Insulin receptor structure and its implications for the IGF-1 receptor. Curr Opin Struct Biol 2007;17:699-705. ArticlePubMed
- 18. Myers MG Jr, White MF. Insulin signal transduction and the IRS proteins. Annu Rev Pharmacol Toxicol 1996;36:615-658. ArticlePubMed
- 19. Bandyopadhyay G, Standaert ML, Zhao L, Yu B, Avignon A, Galloway L, Karnam P, Moscat J, Farese RV. Activation of protein kinase C (alpha, beta, and zeta) by insulin in 3T3/L1 cells. Transfection studies suggest a role for PKC-zeta in glucose transport. J Biol Chem 1997;272:2551-2558. PubMed
- 20. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012;55:2565-2582. ArticlePubMedPMCPDF
- 21. Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A 2003;100:7265-7270. ArticlePubMedPMC
- 22. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796-1808. ArticlePubMedPMC
- 23. Sutton JS, Weiss L. Transformation of monocytes in tissue culture into macrophages, epithelioid cells, and multinucleated giant cells. An electron microscope study. J Cell Biol 1966;28:303-332. PubMedPMC
- 24. Gouon-Evans V, Rothenberg ME, Pollard JW. Postnatal mammary gland development requires macrophages and eosinophils. Development 2000;127:2269-2282. ArticlePubMedPDF
- 25. Banaei-Bouchareb L, Peuchmaur M, Czernichow P, Polak M. A transient microenvironment loaded mainly with macrophages in the early developing human pancreas. J Endocrinol 2006;188:467-480. ArticlePubMed
- 26. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175-184. ArticlePubMedPMC
- 27. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009;27:451-483. ArticlePubMed
- 28. Chait A, Kim F. Saturated fatty acids and inflammation: who pays the toll? Arterioscler Thromb Vasc Biol 2010;30:692-693. ArticlePubMed
- 29. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 2006;116:115-124. ArticlePubMed
- 30. Kewalramani G, Fink LN, Asadi F, Klip A. Palmitate-activated macrophages confer insulin resistance to muscle cells by a mechanism involving protein kinase C θ and ε. PLoS One 2011;6:e26947ArticlePubMedPMC
- 31. Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 2007;282:35279-35292. ArticlePubMed
- 32. Fernandez-Veledo S, Nieto-Vazquez I, Vila-Bedmar R, Garcia-Guerra L, Alonso-Chamorro M, Lorenzo M. Molecular mechanisms involved in obesity-associated insulin resistance: therapeutical approach. Arch Physiol Biochem 2009;115:227-239. ArticlePubMed
- 33. Plomgaard P, Nielsen AR, Fischer CP, Mortensen OH, Broholm C, Penkowa M, Krogh-Madsen R, Erikstrup C, Lindegaard B, Petersen AM, Taudorf S, Pedersen BK. Associations between insulin resistance and TNF-alpha in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia 2007;50:2562-2571. ArticlePubMedPDF
- 34. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997;389:610-614. ArticlePubMedPDF
- 35. Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 2003;278:45777-45784. PubMed
- 36. Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 2002;51:3391-3399. ArticlePubMedPDF
- 37. Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 2001;357(Pt 3):593-615. ArticlePubMedPMCPDF
- 38. Tsuchiya K, Sakai H, Suzuki N, Iwashima F, Yoshimoto T, Shichiri M, Hirata Y. Chronic blockade of nitric oxide synthesis reduces adiposity and improves insulin resistance in high fat-induced obese mice. Endocrinology 2007;148:4548-4556. ArticlePubMedPDF
- 39. Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 2001;7:1138-1143. ArticlePubMedPDF
- 40. Becerril S, Rodriguez A, Catalan V, Sainz N, Ramirez B, Collantes M, Penuelas I, Gomez-Ambrosi J, Fruhbeck G. Deletion of inducible nitric-oxide synthase in leptin-deficient mice improves brown adipose tissue function. PLoS One 2010;5:e10962ArticlePubMedPMC
- 41. Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 2005;437:759-763. ArticlePubMedPMCPDF
- 42. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPAR-gamma controls alternative activation and improves insulin resistance. Nature 2007;447:1116-1120. ArticlePubMedPMCPDF
- 43. Rieusset J, Auwerx J, Vidal H. Regulation of gene expression by activation of the peroxisome proliferator-activated receptor gamma with rosiglitazone (BRL 49653) in human adipocytes. Biochem Biophys Res Commun 1999;265:265-271. PubMed
- 44. Kramer D, Shapiro R, Adler A, Bush E, Rondinone CM. Insulin-sensitizing effect of rosiglitazone (BRL-49653) by regulation of glucose transporters in muscle and fat of Zucker rats. Metabolism 2001;50:1294-1300. ArticlePubMed
- 45. Martens FM, Visseren FL, Lemay J, de Koning EJ, Rabelink TJ. Metabolic and additional vascular effects of thiazolidinediones. Drugs 2002;62:1463-1480. ArticlePubMed
- 46. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821-1830. ArticlePubMedPMC
- 47. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 2011;480:104-108. ArticlePubMedPMCPDF
- 48. Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL. Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology 2002;143:2376-2384. ArticlePubMed
- 49. Stunes AK, Westbroek I, Gustafsson BI, Fossmark R, Waarsing JH, Eriksen EF, Petzold C, Reseland JE, Syversen U. The peroxisome proliferator-activated receptor (PPAR) alpha agonist fenofibrate maintains bone mass, while the PPAR gamma agonist pioglitazone exaggerates bone loss, in ovariectomized rats. BMC Endocr Disord 2011;11:11ArticlePubMedPMCPDF
- 50. Okazaki R, Miura M, Toriumi M, Taguchi M, Hirota Y, Fukumoto S, Fujita T, Tanaka K, Takeuchi A. Short-term treatment with troglitazone decreases bone turnover in patients with type 2 diabetes mellitus. Endocr J 1999;46:795-801. ArticlePubMed
- 51. Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA, Park SW, Lane NE, Harris TB, Cummings SR. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab 2006;91:3349-3354. ArticlePubMed
- 52. Raskin P, Rendell M, Riddle MC, Dole JF, Freed MI, Rosenstock J. Rosiglitazone Clinical Trials Study Group. A randomized trial of rosiglitazone therapy in patients with inadequately controlled insulin-treated type 2 diabetes. Diabetes Care 2001;24:1226-1232. ArticlePubMedPDF
- 53. Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, Le Winter M, Porte D, Semenkovich CF, Smith S, Young LH, Kahn R. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004;27:256-263. ArticlePubMedPDF
- 54. Sena S, Rasmussen IR, Wende AR, McQueen AP, Theobald HA, Wilde N, Pereira RO, Litwin SE, Berger JP, Abel ED. Cardiac hypertrophy caused by peroxisome proliferator-activated receptor-gamma agonist treatment occurs independently of changes in myocardial insulin signaling. Endocrinology 2007;148:6047-6053. PubMed
- 55. Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T. SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res 2010;38:7458-7471. PubMedPMC
- 56. Wang C, Powell MJ, Popov VM, Pestell RG. Acetylation in nuclear receptor signaling and the role of sirtuins. Mol Endocrinol 2008;22:539-545. ArticlePubMedPDF
- 57. Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010;466:451-456. ArticlePubMedPMCPDF
- 58. Kamenecka TM, Busby SA, Kumar N, Choi JH, Banks AS, Vidovic D, Cameron MD, Schurer SC, Mercer BA, Hodder P, Spiegelman BM, Griffin PR. Potent anti-diabetic actions of a novel non-agonist PPARgamma ligand that blocks Cdk5-mediated phosphorylation updated 2013 Mar 7. Available from: http://www.ncbi.nlm.nih.gov/books/NBK143191/pdf/ml244.pdf.
- 59. Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schurer SC, Vidovic D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 2011;477:477-481. ArticlePubMedPMCPDF
REFERENCES
Fig. 1Anticipated regulatory effects of selective peroxisome proliferator-activated receptor-γ (PPAR-γ) antagonists in adipocytes and macrophages: improved expression of insulin-sensitiziting adipokines and lessened inflammatory response. TLR, Toll-like receptor; TNF, tumor necrosis factor; IL-6, interleukin-6; NO, nitric oxide.
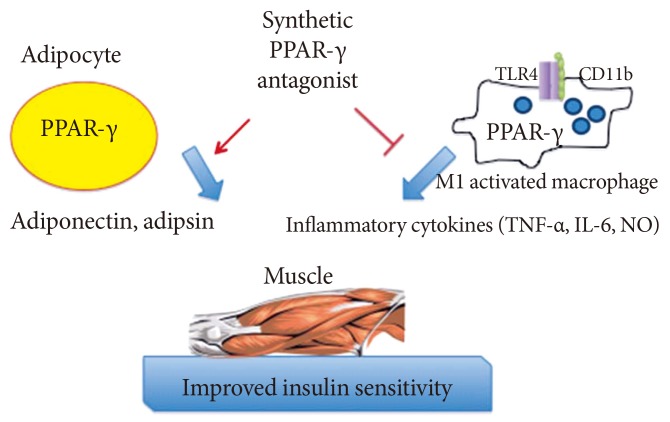
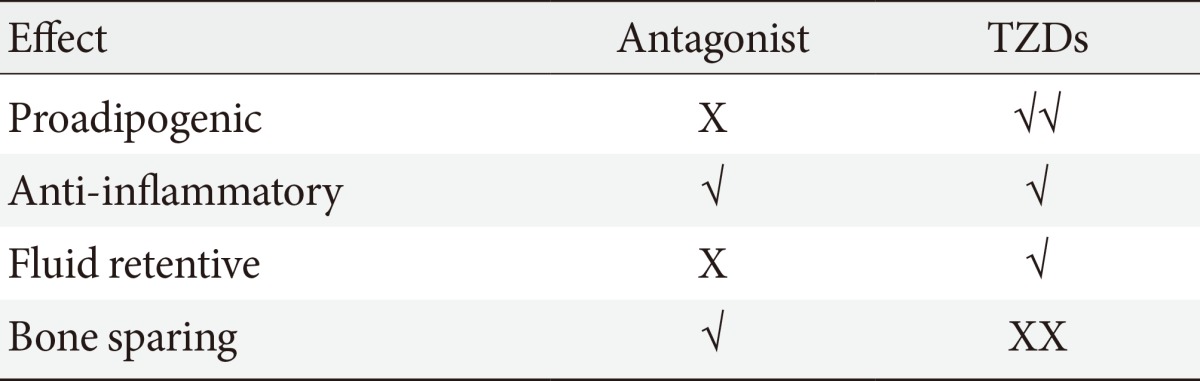
Figure & Data
References
Citations
Citations to this article as recorded by 
- Lysine 222 in PPAR γ1 functions as the key site of MuRF2-mediated ubiquitination modification
Yucheng Fan, Fangjing Xu, Rui Wang, Jun He
Scientific Reports.2023;[Epub] CrossRef - Heart failure and diabetes: Clinical significance and epidemiology of this two‐way association
Terri Jerkins, Janet B. McGill, David S. H. Bell
Diabetes, Obesity and Metabolism.2023; 25(S3): 3. CrossRef - The pleiotropic peroxisome proliferator activated receptors: Regulation and therapeutics
Gargi Dixit, Arati Prabhu
Experimental and Molecular Pathology.2022; 124: 104723. CrossRef - The Effect of PPARγ rs1801282 Variant on Mortality Risk Among Asians With Chronic Kidney Disease: A Cohort Study and Meta-Analysis
Wei-Teing Chen, Chih-Chien Chiu, Dung-Jang Tsai, Pi-Shao Ko, Meng-Chang Lee, Hsiao-Ting Lin, Ying-Kai Chen, Wen Su, Yuh-Feng Lin, Sui-Lung Su
Frontiers in Genetics.2022;[Epub] CrossRef - Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From NAFLD to NASH to HCC
Hyunmi Kim, Da Som Lee, Tae Hyeon An, Hyun-Ju Park, Won Kon Kim, Kwang-Hee Bae, Kyoung-Jin Oh
International Journal of Molecular Sciences.2021; 22(9): 4495. CrossRef - Associations between obesity-related gene expression in maternal and cord blood and newborn adiposity: findings from the Araraquara Cohort study
P. Nakandakare, C. F. Nicoletti, N. Y. Noronha, C. B. Nonino, P. P. Argentato, N. N. Dejani, L. A. Luzia, M. M. Rogero, P. H. C. Rondó
International Journal of Obesity.2021; 45(9): 1958. CrossRef - PPARG Pro12Ala Polymorphism with CKD in Asians: A Meta-Analysis Combined with a Case-Control Study—A Key for Reaching Null Association
Hsiang-Cheng Chen, Wei-Teing Chen, Tzu-Ling Sung, Dung-Jang Tsai, Chin Lin, Hao Su, Yuh-Feng Lin, Hung-Yi Chiu, Sui-Lung Su
Genes.2020; 11(6): 705. CrossRef - Induction of peroxisome proliferator activated receptor γ (PPARγ) mediated gene expression and inhibition of induced nitric oxide production by Maerua subcordata (Gilg) DeWolf
Mebrahtom Gebrelibanos Hiben, Laura de Haan, Bert Spenkelink, Sebastiaan Wesseling, Jacques Vervoort, Ivonne M. C. M. Rietjens
BMC Complementary Medicine and Therapies.2020;[Epub] CrossRef - UPR modulation of host immunity by Pseudomonas aeruginosa in cystic fibrosis
Brahmchetna Bedi, Kuo-Chuan. Lin, Nicholas M. Maurice, Zhihong Yuan, Kaiser Bijli, Michael Koval, C. Michael Hart, Joanna B. Goldberg, Arlene Stecenko, Ruxana T. Sadikot
Clinical Science.2020; 134(14): 1911. CrossRef - Sepsis Immunometabolism: From Defining Sepsis to Understanding How Energy Production Affects Immune Response
Ioannis Koutroulis, Rachael Batabyal, Brittany McNamara, Matthew Ledda, Claire Hoptay, Robert J. Freishtat
Critical Care Explorations.2019; 1(11): e0061. CrossRef - Combining SGLT2 Inhibition With a Thiazolidinedione Additively Attenuate the Very Early Phase of Diabetic Nephropathy Progression in Type 2 Diabetes Mellitus
Eugene Han, Eugene Shin, Gyuri Kim, Ji-Yeon Lee, Yong-ho Lee, Byung-Wan Lee, Eun Seok Kang, Bong-Soo Cha
Frontiers in Endocrinology.2018;[Epub] CrossRef - Role of oral hypoglycemic drugs on inflammatory condition associated with type 2 diabetes mellitus
Shamim Shaikh Mohiuddin
Journal of Diabetes, Metabolic Disorders & Control.2018; 5(2): 78. CrossRef - Effects of lobeglitazone on insulin resistance and hepatic steatosis in high-fat diet-fed mice
Bong-Hoi Choi, Zhen Jin, Chin-ok Yi, Juhong Oh, Eun Ae Jeong, Jong Youl Lee, Kyung-ah Park, Kyung Eun Kim, Jung Eun Lee, Hyun-Jin Kim, Jong Ryeal Hahm, Gu Seob Roh, Jonathan M Peterson
PLOS ONE.2018; 13(7): e0200336. CrossRef - Cucurbita ficifolia (Cucurbitaceae) modulates inflammatory cytokines and IFN-γ in obese mice
Á. Fortis-Barrera, R. García-Macedo, J.C. Almanza-Perez, G. Blancas-Flores, A. Zamilpa-Alvarez, J.L. Flores-Sáenz, M. Cruz, R. Román-Ramos, F.J. Alarcón-Aguilar
Canadian Journal of Physiology and Pharmacology.2017; 95(2): 170. CrossRef - Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness
Yong-ho Lee, Jae Hyeon Kim, So Ra Kim, Heung Yong Jin, Eun-Jung Rhee, Young Min Cho, Byung-Wan Lee
Journal of Korean Medical Science.2017; 32(1): 60. CrossRef -
Peroxisome proliferator‐activated receptor‐γ agonists attenuate biofilm formation by
Pseudomonas aeruginosa
Brahmchetna Bedi, Nicholas M. Maurice, Vincent T. Ciavatta, K. Sabrina Lynn, Zhihong Yuan, Samuel A. Molina, Myungsoo Joo, William R. Tyor, Joanna B. Goldberg, Michael Koval, C. Michael Hart, Ruxana T. Sadikot
The FASEB Journal.2017; 31(8): 3608. CrossRef - Pioglitazone and the secondary prevention of cardiovascular disease. A meta-analysis of randomized-controlled trials
Marit de Jong, H. Bart van der Worp, Yolanda van der Graaf, Frank L. J. Visseren, Jan Westerink
Cardiovascular Diabetology.2017;[Epub] CrossRef - Gestational diabetes mellitus was related to ambient air pollutant nitric oxide during early gestation
Shih-Chun Pan, Ching-Chun Huang, Shio-Jean Lin, Bing-Yu Chen, Chang-Chuan Chan, Yue-Liang Leon Guo
Environmental Research.2017; 158: 318. CrossRef - The Multifaceted Haptoglobin in the Context of Adipose Tissue and Metabolism
Margherita Maffei, Ilaria Barone, Gaia Scabia, Ferruccio Santini
Endocrine Reviews.2016; 37(4): 403. CrossRef - Enhanced Clearance of Pseudomonas aeruginosa by Peroxisome Proliferator-Activated Receptor Gamma
Brahmchetna Bedi, Zhihong Yuan, Myungsoo Joo, Susu M. Zughaier, Joanna B. Goldberg, Jack L. Arbiser, C. Michael Hart, Ruxana T. Sadikot, B. A. McCormick
Infection and Immunity.2016; 84(7): 1975. CrossRef - F-box only protein 9 is an E3 ubiquitin ligase of PPARγ
Kyeong Won Lee, Soo Heon Kwak, Young Do Koo, Yun-Kyung Cho, Hak Mo Lee, Hye Seung Jung, Young Min Cho, Young Joo Park, Sung Soo Chung, Kyong Soo Park
Experimental & Molecular Medicine.2016; 48(5): e234. CrossRef - The Resin fromProtium heptaphyllumPrevents High-Fat Diet-Induced Obesity in Mice: Scientific Evidence and Potential Mechanisms
Karine Maria Martins Bezerra Carvalho, José Delano Barreto Marinho Filho, Tiago Sousa de Melo, Ana Jérsia Araújo, Josiane da Silva Quetz, Maria do Perpétuo Socorro Saldanha da Cunha, Karina Moura de Melo, Armenio Andre de Carvalho Almeida da Silva, Adrian
Evidence-Based Complementary and Alternative Medicine.2015; 2015: 1. CrossRef - Lobeglitazone and pioglitazone as add‐ons to metformin for patients with type 2 diabetes: a 24‐week, multicentre, randomized, double‐blind, parallel‐group, active‐controlled, phase III clinical trial with a 28‐week extension
S.‐M. Jin, C.‐Y. Park, Y. M. Cho, B. J. Ku, C. W. Ahn, B.‐S. Cha, K. W. Min, Y. A. Sung, S. H. Baik, K. W. Lee, K.‐H. Yoon, M.‐K. Lee, S. W. Park
Diabetes, Obesity and Metabolism.2015; 17(6): 599. CrossRef - Deconvolution of Complex 1D NMR Spectra Using Objective Model Selection
Travis S. Hughes, Henry D. Wilson, Ian Mitchelle S. de Vera, Douglas J. Kojetin, Paul C. Driscoll
PLOS ONE.2015; 10(8): e0134474. CrossRef - PPARγ partial agonist GQ-16 strongly represses a subset of genes in 3T3-L1 adipocytes
Flora Aparecida Milton, Aleksandra Cvoro, Angelica A. Amato, Douglas H. Sieglaff, Carly S. Filgueira, Anithachristy Sigamani Arumanayagam, Maria do Carmo Alves de Lima, Ivan Rocha Pitta, Francisco de Assis Rocha Neves, Paul Webb
Biochemical and Biophysical Research Communications.2015; 464(3): 718. CrossRef - Voluntary exercise prevents colonic inflammation in high-fat diet-induced obese mice by up-regulating PPAR-γ activity
Wei-Xin Liu, Ting Wang, Feng Zhou, Ying Wang, Jun-Wei Xing, Shen Zhang, Shou-Zhi Gu, Li-Xuan Sang, Cong Dai, Hai-Lan Wang
Biochemical and Biophysical Research Communications.2015; 459(3): 475. CrossRef - Effect of a new PPAR-gamma agonist, lobeglitazone, on neointimal formation after balloon injury in rats and the development of atherosclerosis
Soo Lim, Kuy-Sook Lee, Jie Eun Lee, Ho Seon Park, Kyoung Min Kim, Jae Hoon Moon, Sung Hee Choi, Kyong Soo Park, Young Bum Kim, Hak Chul Jang
Atherosclerosis.2015; 243(1): 107. CrossRef - Antidiabetic agents: Potential anti-inflammatory activity beyond glucose control
A.J. Scheen, N. Esser, N. Paquot
Diabetes & Metabolism.2015; 41(3): 183. CrossRef - Genomic binding and regulation of gene expression by the thyroid carcinoma-associated PAX8-PPARG fusion protein
Yanxiao Zhang, Jingcheng Yu, Chee Lee, Bin Xu, Maureen A. Sartor, Ronald J. Koenig
Oncotarget.2015; 6(38): 40418. CrossRef - Peroxisome Proliferator-Activated Receptor γ (PPARγ) and Ligand Choreography: Newcomers Take the Stage
Santiago Garcia-Vallvé, Laura Guasch, Sarah Tomas-Hernández, Josep Maria del Bas, Vincent Ollendorff, Lluís Arola, Gerard Pujadas, Miquel Mulero
Journal of Medicinal Chemistry.2015; 58(14): 5381. CrossRef - Pathway Analysis of Metabolic Syndrome Using a Genome-Wide Association Study of Korea Associated Resource (KARE) Cohorts
Unjin Shim, Han-Na Kim, Yeon-Ah Sung, Hyung-Lae Kim
Genomics & Informatics.2014; 12(4): 195. CrossRef - Pax-8–PPAR-γ fusion protein in thyroid carcinoma
Priyadarshini Raman, Ronald J. Koenig
Nature Reviews Endocrinology.2014; 10(10): 616. CrossRef - Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients: a systemic review and meta-analysis
Shinan Yin, Hua Bai, Danqing Jing
Diagnostic Pathology.2014;[Epub] CrossRef - Common biological mechanisms between bipolar disorder and type 2 diabetes: Focus on inflammation
Ajaykumar N. Sharma, Isabelle E. Bauer, Marsal Sanches, Juan F. Galvez, Giovana B. Zunta-Soares, Joao Quevedo, Flavio Kapczinski, Jair C. Soares
Progress in Neuro-Psychopharmacology and Biological Psychiatry.2014; 54: 289. CrossRef
 PubReader
PubReader Cite
Cite