- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 36(6); 2012 > Article
-
ReviewOthers Periodontitis and Insulin Resistance: Casual or Causal Relationship?
- Abhijit N. Gurav
-
Diabetes & Metabolism Journal 2012;36(6):404-411.
DOI: https://doi.org/10.4093/dmj.2012.36.6.404
Published online: December 12, 2012
Department of Periodontics, Tatyasaheb Kore Dental College & Research Centre, Kolhapur, India.
- Corresponding author: Abhijit N. Gurav. Department of Periodontics, Tatyasaheb Kore Dental College & Research Centre, New Pargaon, Kolhapur-416137, India. drabhijitgurav@gmail.com
Copyright © 2012 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Insulin resistance (IR) is now considered as a chronic and low level inflammatory condition. It is closely related to altered glucose tolerance, hypertriglyceridemia, abdominal obesity, and coronary heart disease. IR is accompanied by the increase in the levels of inflammatory cytokines like interleukin-1 and 6, tumor necrosis factor-α. These inflammatory cytokines also play a crucial part in pathogenesis and progression of insulin resistance. Periodontitis is the commonest of oral diseases, affecting tooth investing tissues. Pro-inflammatory cytokines are released in the disease process of periodontitis. Periodontitis can be attributed with exacerbation of IR. Data in the literature supports a "two way relationship" between diabetes and periodontitis. Periodontitis is asymptomatic in the initial stages of disease process and it often escapes diagnosis. This review presents the blurred nexus between periodontitis and IR, underlining the pathophysiology of the insidious link. The knowledge of the association between periodontitis and IR can be valuable in planning effectual treatment modalities for subjects with altered glucose homeostasis and diabetics. Presently, the studies supporting this association are miniscule. Further studies are mandatory to substantiate the role of periodontitis in the deterioration of IR.
- Diabetes mellitus (DM) is a scourge to the global community, stepping up at a magnanimous proportion. As per the data provided by International Diabetes Federation, the worldwide prevalence of DM in 2011 was 366 million and this number is projected to reach 552 million by 2030 [1]. Type 2 diabetes mellitus (T2DM) follows as a result of impaired glucose homeostasis. Insulin resistance (IR) is measured by glucose homeostasis model assessment (HOMA), first described by Matthews and colleagues [2]. T2DM is characterized as a non-autoimmune condition, which involves multiple, intriguing factors like genetics, environmental or acquired factors and presence of inflammatory pathways. IR is a complicated condition involving multiple etiological pathways. It plays a crucial part in the pathogenesis of metabolic syndrome and T2DM, yet the inherent mechanisms are not completely cognizant [3]. Periodontitis is the most common oral infection with wide global prevalence. Clinical features of periodontitis include bleeding gingiva, increased interdental spacing, increase in probing depth, bad oral breath and mobility of teeth in advanced cases. Since periodontitis is asymptomatic, the affected subjects are largely unaware and refrain from periodontal treatment. Periodontitis is characterized by the loss of tooth supporting tissues, which is indolent in nature with marked chronicity. The primary etiology of periodontitis is dental plaque, which houses multiple bacteria of different strains and species [4]. It has been proven that periodontitis has effects, impacting the systemic health of the subject and the detrimental effects are not only confined to the oral cavity [5]. Periodontal medicine is an emerging branch which addresses the various links of periodontitis with systemic diseases. Periodontitis DM maintain a "two way relationship" [6]. The present review addresses the issue of IR in particular and the potential causal role of periodontitis in pathogenesis of the same.
INTRODUCTION
- Periodontitis is essentially a biofilm induced disease, initiated and progressed by different bacterial species, present in the dental plaque. The periodontopathic bacteria are basically gram-negative in nature and they are present in the depths of periodontal pockets, placed at low oxygen tension. The putative pathogenic bacteria express noxious toxins instrumental for the periodontal destruction [7]. Currently, the consensus regarding pathogenesis of periodontitis has undergone an immense change. According to this concept, periodontitis is not only the result of adverse microbial activity but as an interaction among various other factors like genetics, systemic health, immunity, environmental factors like tobacco and stress. The above mentioned factors play an important role in the modification of host response to the disease process. Thus, sometimes the periodontal disease may exhibit varied expression [7]. Various pro-inflammatory mediators like interleukin (IL)-1α and IL-1β, IL-6, tumor necrosis factor (TNF)-α, prostaglandin E2 (PGE2), matrix metalloproteinases are expressed in periodontitis, as a result of activation of the host immune-inflammatory mechanisms. Cytokines are liberated by periodontal tissues like fibroblasts, endothelial cells, macrophages, osteoclasts, epithelial cells, neutrophils, monocytes, lymphocytes, and mast cells. Immune cells like neutrophils, monocytes also let out cytokines in inflammatory conditions. This host tissue expressed array of factors may be detrimental to the host tissue itself, amplifying the destructive disease process [8]. The periodontopathogenic flora produce toxins and significant challenge is offered by lipopolysaccharide (LPS), a component of the gram-negative bacterial cell wall. LPS is a potent endotoxin which exacerbates the host inflammatory response. Subjects with periodontitis are reported to present endotoxin activity in the serum [9]. As discussed previously the bacteria are amicably housed in the periodontal pocket. These bacteria, attended with their noxious products can gain a ready access through the ulcerated lining of the periodontal pocket, into the systemic circulation. Loos [10] reported a significant cumulative surface area of all periodontal lesions in a patient with severe periodontitis, ranging from 15 to 20 cm2. Further, the periodontal inflamed surface area (PISA) can be used as a tool to accurately assess the amount of periodontal inflamed tissue in a subject with periodontitis [11]. Thus, it can be inferred that, in severe periodontitis patients, pro-inflammatory mediators (IL-1α and IL-1β, TNF-α, PGE2) from the disease gingival sites may be 'poured' into the systemic circulation. Studies have identified many systemic biomarkers, exposing the link of periodontitis with systemic conditions and cardiovascular disease [12,13]. Thus, it can be enunciated that periodontitis is a "low grade infection" capable of developing a "low grade systemic inflammation" with an ability to influence the general systemic health.
ETIOPATHOGENESIS OF PERIODONTITIS AND ITS SYSTEMIC LINK
- IR, a precursor to T2DM has a complicated metabolic mechanism, with multiple etiological pathways. It is proposed that a defect in insulin receptor substrate (IRS) protein function is necessary for the uncoupling of the insulin signal, resulting in IR [14]. Various protein kinases, which are important in insulin signaling, are key players in IR [15]. Insulin functions by binding to the heterotetrameric membrane receptor leading to IRS-1 phosphorylation and IRS-1-associated phosphatidylinositol 3 phosphate kinase (PI3 kinase) activation [16]. This event in turn impacts effectors like Akt/protein kinase B (PKB), which triggers the glucose transporter GLUT4. GLUT4 is further translocated into the membrane and induces glucose import into the cell [17]. Protein kinase C (PKC) isoenzymes is a family of signaling molecules involved in the actions of insulin. These PKCs are categorized as classical isoenzymes, novel isoenzymes, and atypical isoenzymes [18]. They operate in an elaborate manner and are noted to play a positive and negative regulatory role in insulin signaling [19,20]. Signal activity from a stimulus to the ordinance of cellular processes, considering those involved in glucose homeostsis, mainly depends upon protein kinase signaling. A defect in the insulin signaling process results in downregulation of Akt/PKB, thereby inhibiting the required cascade process. Kinases like Jun N-terminal kinases (JNKs), also named SAPKs can phosphorylate IRS-1 & 2 at specific serine and threonine residues, leading to suppression of insulin signaling [21]. Peroxisome proliferator-activated receptor (PPAR)-γ complements insulin signaling. Pro-inflammatory cytokines increase the IR, by activation of JNK and IκB kinase-β/nuclear factor κB and downregulation of PPAR-γ [22,23].
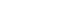
- A "bidirectional relationship" between periodontitis and DM has been proposed (Fig. 1). It is theorized that pro-inflammatory cytokines expressed by gingiva in periodontitis enter the systemic circulation leading to exacerbation of DM. Conversely, the elevated levels of the pro-inflammatory cytokines in DM may reach the gingiva leading to aggravation of already existing periodontal disease [24,25]. Chronic subclinical systemic inflammation may be conducive to impaired glucose homeostasis/increased IR, subsequently paving a way for clinical manifestation of T2DM [23].
- Chronic hyperglycemia in T2DM, indulges nonenzymatic glycation of proteins with the formation of advanced glycation end products (AGEs), which are reported to prime the macrophages to express cytokines (IL-6 and TNF-α). These cytokines are instrumental in the release of acute phase reactants (CRP) from the liver, further amplifying the existing inflammation [26]. Currently, adipose tissue is regarded as an endocrine organ, a major depot, capable of secreting bioactive agents called adipokines [27]. Adipokines enlisted in regulation of IR are adiponectin, leptin, resistin, visfatin, chemerin, TNF-α, IL-1, IL-6, IL-8, IL-10, plasminogen-aktivator-inhibitor-1, monocyte chemoattractant protein-1, and retinol binding protein-4 [28]. Adiponectin maintains a mutual antagonistic action to TNF-α, plays an important role as anti-diabetic, anti-atherogenic, anti-inflammatory agent. Studies report that TNF-α inhibits the expression of adiponectin. Conversely, adiponectin suppresses LPS induced TNF-α production [29,30]. TNF-α is one of the most important cytokine implicated in the initiation and progression of IR.
- Table 1 depicts the possible mechanisms by which TNF-α contributes to IR [31-38]. The role of IL-6 in IR is controversial as reported in the literature [39,40]. It can be inferred that relentless increase in the systemic levels of IL-6, as in obesity and T2DM may lead to IR, whereas a transient increase in IL-6 may assist in normal glucose homeostasis [41]. With the new entropy in the advancement of molecular biology an increased onus is laid upon the role of inflammatory mediators in the pathogenesis of IR and subsequent T2DM. Periodontitis and T2DM share a common process of pathogenesis, involving inflammatory response at the local and systemic level [42]. Studies directed at assessing the IR utilize HOMA for estimation of insulin sensitivity. The HOMA method deduces an estimate of insulin sensitivity from a mathematical model of fasting plasma glucose and insulin concentrations [2].
- Table 2 outlines the studies relating IR with periodontitis. The studies depicted in the table are animal and human studies [43-49]. Obesity has also been portrayed as a condition associated with low grade systemic inflammation, demonstrating commonality in the expression of identical cytokines as that detected in periodontitis and T2DM [46]. T2DM subjects with concomitant periodontitis exhibit increased biomarkers and oxidative stress. Compromised β-cell function and increased IR is attributed to the intensified oxidative stress as a result of hyperactivated neutrophils in periodontitis, ensuing in the boosted release of reactive oxygen species [49,50].
- A common denominator that exists for periodontitis and T2DM is the systemic presence of common pro-inflammatory cytokines. Obesity is now believed to bear a causal relationship with periodontitis [51,52]. A bidirectional association between DM and periodontitis has been emphasized by abundant literature [6,53,54]. It is proposed that periodontitis and IR are potential risk factors for the perturbation of cardiovascular health [55-57]. IR is an important component of metabolic syndrome and it is proposed that periodontal disease should be considered as a component of metabolic syndrome [42,58]. It is important to acknowledge periodontitis, as an emerging risk factor for metabolic syndrome. There is still a sizeable vacuum in evidence based literature, with regards to the connection of periodontitis and IR. This issue has not been adequately addressed in majority of the studies. The studies in rodent models are worthy for realizing the potential cellular mechanisms of the pathogenesis with IR, but there is still doubt whether the pathways and trials in these animal models can be extrapolated in humans. The PISA contributes as a dynamic source for the progression of poor metabolic control in T2DM subjects with severe periodontitis. Nesse et al. [59] have calculated that an increase of PISA with 333 mm2 was associated with a 1.0 percentage point augmentation of HbA1c, independent of other factors. A similar study should be conducted which can indicate a dose-response relationship between PISA and IR. PISA can serve as a valuable tool to quantify the inflammatory burden in periodontitis and relate it with IR.
ETIOPATHOGENESIS OF IR AND POTENTIAL LINK WITH PERIODONTITIS
- Pro-inflammatory cytokines amplify IR. IR may be a constituent of the causal pathway connecting inflammatory mediators to incident diabetes. There is a lack of research in human subjects concerning periodontitis, as a causal pathology for IR. Periodontitis and IR are largely unrecognized. Hence it is connoted, to perceive the early presence of IR and periodontitis. Both the conditions existing conjointly in the same individual, can reciprocate the pernicious effects of each other. Extensive, multicentric, randomized controlled trials involving large populations are vindicated to analyze the elusive link between periodontitis and IR. The potential effects of periodontal therapy in the de-escalation of IR should be contemplated. The potential favorable benefits of anti-cytokine therapy to treat IR should be explored. Although, the high prevalence of periodontitis in diabetics is cognizant by the dental professionals, it is not a well known fact in the medical community. Periodontitis should receive due attention as a "pandemic" by the respective national and world health governance. Both, periodontitis and DM, are cryptically linked to metabolic syndrome and cardiovascular diseases. The medical and oral health professional should align efforts in management of T2DM susceptible subjects with periodontitis. Concerted endeavors can be valuable to control the progression of both the conditions respectively.
CONCLUSIONS
- 1. Lam DW, LeRoith D. The worldwide diabetes epidemic. Curr Opin Endocrinol Diabetes Obes 2012;19:93-96. ArticlePubMed
- 2. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412-419. ArticlePubMedPDF
- 3. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012;148:852-871. ArticlePubMedPMC
- 4. Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet 2005;366:1809-1820. ArticlePubMed
- 5. Williams RC, Barnett AH, Claffey N, Davis M, Gadsby R, Kellett M, Lip GY, Thackray S. The potential impact of periodontal disease on general health: a consensus view. Curr Med Res Opin 2008;24:1635-1643. ArticlePubMed
- 6. Grossi SG, Genco RJ. Periodontal disease and diabetes mellitus: a two-way relationship. Ann Periodontol 1998;3:51-61. ArticlePubMed
- 7. Benakanakere M, Kinane DF. Innate cellular responses to the periodontal biofilm. Front Oral Biol 2012;15:41-55. ArticlePubMed
- 8. Preshaw PM, Taylor JJ. How has research into cytokine interactions and their role in driving immune responses impacted our understanding of periodontitis? J Clin Periodontol 2011;38(Suppl 11):60-84. ArticlePubMed
- 9. Ebersole JL, Stevens J, Steffen MJ, Dawson Iii D, Novak MJ. Systemic endotoxin levels in chronic indolent periodontal infections. J Periodontal Res 2010;45:1-7. ArticlePubMedPMC
- 10. Loos BG. Systemic markers of inflammation in periodontitis. J Periodontol 2005;76(11 Suppl):2106-2115. Article
- 11. Nesse W, Abbas F, van der Ploeg I, Spijkervet FK, Dijkstra PU, Vissink A. Periodontal inflamed surface area: quantifying inflammatory burden. J Clin Periodontol 2008;35:668-673. ArticlePubMed
- 12. Wu T, Trevisan M, Genco RJ, Falkner KL, Dorn JP, Sempos CT. Examination of the relation between periodontal health status and cardiovascular risk factors: serum total and high density lipoprotein cholesterol, C-reactive protein, and plasma fibrinogen. Am J Epidemiol 2000;151:273-282. ArticlePubMed
- 13. Jin LJ, Wang CY. An update on periodontal infections, systemic inflammatory biomarkers, and cardiovascular disease. Chin J Dent Res 2007;10:7-13.
- 14. Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab 2009;296:E581-E591. ArticlePubMed
- 15. Schultze SM, Hemmings BA, Niessen M, Tschopp O. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev Mol Med 2012;14:e1ArticlePubMed
- 16. Bhattacharya S, Dey D, Roy SS. Molecular mechanism of insulin resistance. J Biosci 2007;32:405-413. ArticlePubMedPDF
- 17. Palmada M, Boehmer C, Akel A, Rajamanickam J, Jeyaraj S, Keller K, Lang F. SGK1 kinase upregulates GLUT1 activity and plasma membrane expression. Diabetes 2006;55:421-427. ArticlePubMedPDF
- 18. Lenz JC, Reusch HP, Albrecht N, Schultz G, Schaefer M. Ca2+-controlled competitive diacylglycerol binding of protein kinase C isoenzymes in living cells. J Cell Biol 2002;159:291-302. ArticlePubMedPMCPDF
- 19. Farese RV. Function and dysfunction of aPKC isoforms for glucose transport in insulin-sensitive and insulin-resistant states. Am J Physiol Endocrinol Metab 2002;283:E1-E11. ArticlePubMed
- 20. Kellerer M, Mushack J, Seffer E, Mischak H, Ullrich A, Haring HU. Protein kinase C isoforms alpha, delta and theta require insulin receptor substrate-1 to inhibit the tyrosine kinase activity of the insulin receptor in human kidney embryonic cells (HEK 293 cells). Diabetologia 1998;41:833-838. ArticlePubMedPDF
- 21. Tarantino G, Caputi A. JNKs, insulin resistance and inflammation: a possible link between NAFLD and coronary artery disease. World J Gastroenterol 2011;17:3785-3794. ArticlePubMedPMC
- 22. Wang T, Villegas S, Huang Y, White SK, Ahlem C, Lu M, Olefsky JM, Reading C, Frincke JM, Alleva D, Flores-Riveros J. Amelioration of glucose intolerance by the synthetic androstene HE3286: link to inflammatory pathways. J Pharmacol Exp Ther 2010;333:70-80. ArticlePubMed
- 23. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793-1801. ArticlePubMedPMC
- 24. Nishimura F, Iwamoto Y, Mineshiba J, Shimizu A, Soga Y, Murayama Y. Periodontal disease and diabetes mellitus: the role of tumor necrosis factor-alpha in a 2-way relationship. J Periodontol 2003;74:97-102. PubMed
- 25. Kim J, Amar S. Periodontal disease and systemic conditions: a bidirectional relationship. Odontology 2006;94:10-21. ArticlePubMedPMCPDF
- 26. Janket SJ, Jones JA, Meurman JH, Baird AE, Van Dyke TE. Oral infection, hyperglycemia, and endothelial dysfunction. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;105:173-179. ArticlePubMed
- 27. Lehr S, Hartwig S, Sell H. Adipokines: a treasure trove for the discovery of biomarkers for metabolic disorders. Proteomics Clin Appl 2012;6:91-101. ArticlePubMedPDF
- 28. Hillenbrand A, Weiss M, Knippschild U, Wolf AM, Huber-Lang M. Sepsis-induced adipokine change with regard to insulin resistance. Int J Inflam 2012;2012:972368ArticlePubMedPMCPDF
- 29. Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 2001;50:2094-2099. PubMed
- 30. Park PH, Huang H, McMullen MR, Mandal P, Sun L, Nagy LE. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-alpha production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J Biol Chem 2008;283:26850-26858. PubMedPMC
- 31. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A 1994;91:4854-4858. ArticlePubMedPMC
- 32. Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res 2007;48:751-762. PubMed
- 33. Grunfeld C, Feingold KR. The metabolic effects of tumor necrosis factor and other cytokines. Biotherapy 1991;3:143-158. ArticlePubMedPDF
- 34. Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med 2006;119(5 Suppl 1):S10-S16. ArticlePubMedPMC
- 35. Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 2003;52:1779-1785. PubMed
- 36. Hajri T, Tao H, Wattacheril J, Marks-Shulman P, Abumrad NN. Regulation of adiponectin production by insulin: interactions with tumor necrosis factor-alpha and interleukin-6. Am J Physiol Endocrinol Metab 2011;300:E350-E360. PubMed
- 37. Long SD, Pekala PH. Regulation of GLUT4 mRNA stability by tumor necrosis factor-alpha: alterations in both protein binding to the 3' untranslated region and initiation of translation. Biochem Biophys Res Commun 1996;220:949-953. PubMed
- 38. Hansen LL, Ikeda Y, Olsen GS, Busch AK, Mosthaf L. Insulin signaling is inhibited by micromolar concentrations of H(2) O(2). Evidence for a role of H(2)O(2) in tumor necrosis factor alpha-mediated insulin resistance. J Biol Chem 1999;274:25078-25084. PubMed
- 39. Pedersen BK, Febbraio MA. Point: Interleukin-6 does have a beneficial role in insulin sensitivity and glucose homeostasis. J Appl Physiol 2007;102:814-816. PubMed
- 40. Mooney RA. Counterpoint: interleukin-6 does not have a beneficial role in insulin sensitivity and glucose homeostasis. J Appl Physiol 2007;102:816-818. ArticlePubMed
- 41. Rabe K, Lehrke M, Parhofer KG, Broedl UC. Adipokines and insulin resistance. Mol Med 2008;14:741-751. ArticlePubMedPMCPDF
- 42. Nishimura F, Soga Y, Iwamoto Y, Kudo C, Murayama Y. Periodontal disease as part of the insulin resistance syndrome in diabetic patients. J Int Acad Periodontol 2005;7:16-20. PubMed
- 43. Watanabe K, Petro BJ, Shlimon AE, Unterman TG. Effect of periodontitis on insulin resistance and the onset of type 2 diabetes mellitus in Zucker diabetic fatty rats. J Periodontol 2008;79:1208-1216. ArticlePubMed
- 44. Ekuni D, Tomofuji T, Irie K, Kasuyama K, Umakoshi M, Azuma T, Tamaki N, Sanbe T, Endo Y, Yamamoto T, Nishida T, Morita M. Effects of periodontitis on aortic insulin resistance in an obese rat model. Lab Invest 2010;90:348-359. ArticlePubMedPDF
- 45. Pontes Andersen CC, Flyvbjerg A, Buschard K, Holmstrup P. Periodontitis is associated with aggravation of prediabetes in Zucker fatty rats. J Periodontol 2007;78:559-565. ArticlePubMed
- 46. Genco RJ, Grossi SG, Ho A, Nishimura F, Murayama Y. A proposed model linking inflammation to obesity, diabetes, and periodontal infections. J Periodontol 2005;76(11 Suppl):2075-2084. Article
- 47. Benguigui C, Bongard V, Ruidavets JB, Chamontin B, Sixou M, Ferrieres J, Amar J. Metabolic syndrome, insulin resistance, and periodontitis: a cross-sectional study in a middle-aged French population. J Clin Periodontol 2010;37:601-608. ArticlePubMed
- 48. Timonen P, Suominen-Taipale L, Jula A, Niskanen M, Knuuttila M, Ylostalo P. Insulin sensitivity and periodontal infection in a non-diabetic, non-smoking adult population. J Clin Periodontol 2011;38:17-24. Article
- 49. Allen EM, Matthews JB, DJ OH, Griffiths HR, Chapple IL. Oxidative and inflammatory status in type 2 diabetes patients with periodontitis. J Clin Periodontol 2011;38:894-901. ArticlePubMed
- 50. Karima M, Kantarci A, Ohira T, Hasturk H, Jones VL, Nam BH, Malabanan A, Trackman PC, Badwey JA, Van Dyke TE. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: association with periodontitis. J Leukoc Biol 2005;78:862-870. ArticlePubMedPMCPDF
- 51. Saxlin T, Suominen-Taipale L, Leiviska J, Jula A, Knuuttila M, Ylostalo P. Role of serum cytokines tumour necrosis factor-alpha and interleukin-6 in the association between body weight and periodontal infection. J Clin Periodontol 2009;36:100-105. PubMed
- 52. Gorman A, Kaye EK, Apovian C, Fung TT, Nunn M, Garcia RI. Overweight and obesity predict time to periodontal disease progression in men. J Clin Periodontol 2012;39:107-114. ArticlePubMed
- 53. Mealey BL, Rose LF. Diabetes mellitus and inflammatory periodontal diseases. Curr Opin Endocrinol Diabetes Obes 2008;15:135-141. ArticlePubMed
- 54. Gurav A, Jadhav V. Periodontitis and risk of diabetes mellitus. J Diabetes 2011;3:21-28. ArticlePubMed
- 55. Fernandez-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr Rev 2003;24:278-301. ArticlePubMed
- 56. Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J Gen Intern Med 2008;23:2079-2086. ArticlePubMedPMCPDF
- 57. Friedewald VE, Kornman KS, Beck JD, Genco R, Goldfine A, Libby P, Offenbacher S, Ridker PM, Van Dyke TE, Roberts WC. American Journal of Cardiology. Journal of Periodontology. The American Journal of Cardiology and Journal of Periodontology editors' consensus: periodontitis and atherosclerotic cardiovascular disease. J Periodontol 2009;80:1021-1032. ArticlePubMed
- 58. Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, Wang H, Eckel RH. The metabolic syndrome. Endocr Rev 2008;29:777-822. ArticlePubMedPMC
- 59. Nesse W, Linde A, Abbas F, Spijkervet FK, Dijkstra PU, de Brabander EC, Gerstenbluth I, Vissink A. Dose-response relationship between periodontal inflamed surface area and HbA1c in type 2 diabetics. J Clin Periodontol 2009;36:295-300. ArticlePubMed
REFERENCES

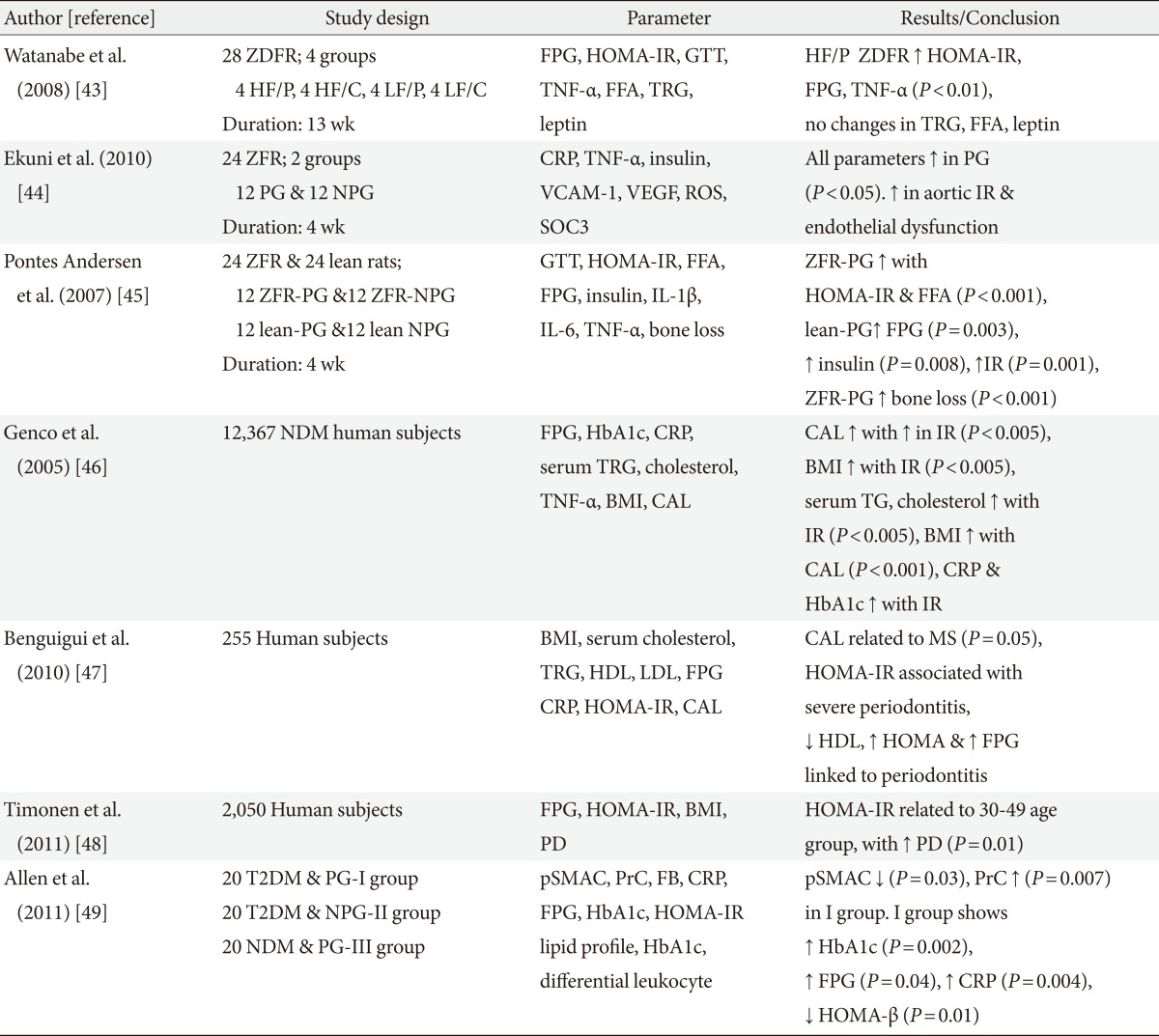
ZDFR, Zucker diabetic fatty rats; HF/P, high fat periodontitis; HF/C, high fat control; LF/P, lean fat periodontitis; LF/C, lean fat control; FPG, fasting plasma glucose; HOMA-IR, homeostasis model assessment of insulin resistance; GTT, glucose tolerance test; TNF-α, tumor necrosis factor-α; FFA, free fatty acids; TRG, triglyceride; ZFR, Zucker fatty rats; PG, periodontitis group; NPG, non-periodontitis group; CRP, C-reactive protein; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; ROS, reactive oxygen species; SOC3, suppressor of cytokine signaling 3; IR, insulin resistance; IL, interleukin; NDM, non-diabetic; HbA1c, glycated hemoglobin; BMI, body mass index; CAL, clinical attachment loss; HDL, high density lipoprotein; LDL, low density lipoprotein; MS, metabolic syndrome; PD, probing depth; T2DM, type 2 diabetes mellitus; pSMAC, plasma small molecule antioxidant capacity; PrC, protein carbonyl; FB, fibrinogen; HOMA-β, homeostasis model assessment of β-cell function; ↓, decreased; ↑, increased.
Figure & Data
References
Citations
- Recent Aspects of Periodontitis and Alzheimer’s Disease—A Narrative Review
Dominika Cichońska, Magda Mazuś, Aida Kusiak
International Journal of Molecular Sciences.2024; 25(5): 2612. CrossRef - Association of Chronic Periodontitis with Helicobacter pylori Infection in Stomach or Mouth: A Systematic Review and Meta-Analysis
Athanasios Tsimpiris, Ioannis Tsolianos, Andreas Grigoriadis, Ioannis Moschos, Dimitrios G. Goulis, Georgios Kouklakis
European Journal of Dentistry.2023; 17(02): 270. CrossRef - Bidirectional association between polycystic ovary syndrome and periodontal diseases
Yang Dou, Jinglei Xin, Peng Zhou, Jianming Tang, Hongliang Xie, Wanting Fan, Zheng Zhang, Donglei Wu
Frontiers in Endocrinology.2023;[Epub] CrossRef - Comparison of the triglyceride glucose index and modified triglyceride glucose indices in assessing periodontitis in Korean adults
Hyun‐Jeong Lee, Ji‐Won Lee, Sue Kim, Yu‐Jin Kwon
Journal of Periodontal Research.2023; 58(3): 503. CrossRef - Risk Assessment of Periodontitis according to Metabolic Score for Insulin Resistance in Korean Population: Korea National Health and Nutrition Examination Survey VI (2013–2015)
In-Hwan Kim, Yea-Chan Lee, Yu-Jin Kwon, Yong-Jae Lee
Korean Journal of Family Practice.2023; 13(1): 38. CrossRef - RELAÇÃO ENTRE OBESIDADE E DOENÇA PERIODONTAL: REVISÃO INTEGRATIVA
Maria Eduarda Ribeiro Da Silva, Karina Sarno Paes Alves Dias, Pedro Gomes Fonseca Rocha, Paollo Teixeira de Amorim Santos, Matheus Feliphe Lima De Melo, Iago Fraga Araújo, Caio Santos Porto, Cícero Gabriel dos Santos Coutinho, Gledyson Sousa Caires
REVISTA FOCO.2023; 16(6): e2257. CrossRef - Non-Insulin-Based Insulin Resistance Indices and Localized Periodontitis in
Physically Active Young Male Adults: CHIEF Oral Health Study
Kun-Zhe Tsai, Yen-Po Lin, Shiue-Wei Lai, Chia-Hsin Liu, Yun-Chen Chang, Gen-Min Lin
Endocrine, Metabolic & Immune Disorders - Drug Targets.2023; 23(7): 937. CrossRef - Periodontal Disease and Its Association with Metabolic Syndrome—A Comprehensive Review
Itay Aizenbud, Asaf Wilensky, Galit Almoznino
International Journal of Molecular Sciences.2023; 24(16): 13011. CrossRef - Periodontitis is associated with insulin resistance in adults living with diabetes mellitus in Uganda: a cross- sectional study
Haruna Muhmood Kiryowa, Ian Guyton Munabi, William Buwembo, Charles Mugisha Rwenyonyi, Erisa Sabakaki Mwaka, Mark Kaddumukasa
BMC Research Notes.2023;[Epub] CrossRef - Periodontal and systemic health of morbidly obese patients eligible for bariatric surgery: a cross-sectional study
Dejana Čolak, Alja Cmok Kučič, Tadeja Pintar, Boris Gašpirc, Rok Gašperšič
BMC Oral Health.2022;[Epub] CrossRef - Impact of Periodontitis on Glycemic Control and Metabolic Status in Diabetes Patients: Current Knowledge on Early Disease Markers and Therapeutic Perspectives
Simona Santonocito, Alessandro Polizzi, Enrico Marchetti, Domenico Dalessandri, Marco Migliorati, Saturnino Marco Lupi, Marco Cicciù, Gaetano Isola, Alessandro Trentini
Mediators of Inflammation.2022; 2022: 1. CrossRef - Do Cytokines Associate Periodontitis with Metabolic Disorders? An
Overview of Current Documents
Reza Aref Nezhad, Hossein Motedayyen, Hossein Roghani-Shahraki
Endocrine, Metabolic & Immune Disorders - Drug Targets.2022; 22(7): 778. CrossRef - Comparison of the triglyceride glucose (TyG) index, triglyceride to high-density lipoprotein cholesterol (TG/HDL-C) ratio, and metabolic score for insulin resistance (METS-IR) associated with periodontitis in Korean adults
Yea-Chan Lee, Ji-Won Lee, Yu-Jin Kwon
Therapeutic Advances in Chronic Disease.2022; 13: 204062232211226. CrossRef - Markers, pathways, and current evidence for periodontitis-associated insulin resistance: A narrative review
VivekKumar Bains, Jaideep Mahendra, Little Mahendra, Madhukar Mittal, Gunam Valli
Journal of International Society of Preventive and Community Dentistry.2022; 12(5): 475. CrossRef - Periodontal Therapy in Bariatric Surgery Patients with Periodontitis: Randomized Control Clinical Trial
Dejana Čolak, Alja Cmok Kučič, Tadeja Pintar, Rok Gašperšič
Journal of Clinical Medicine.2022; 11(22): 6837. CrossRef - Systemic Diseases and Biological Dental Implant Complications: A Narrative Review
Luca Sbricoli, Elissar Bazzi, Edoardo Stellini, Christian Bacci
Dentistry Journal.2022; 11(1): 10. CrossRef - Association Between Diabetic Retinopathy and Periodontitis—A Systematic Review
María Olimpia Paz Alvarenga, Giza Hellen Nonato Miranda, Railson Oliveira Ferreira, Miki Taketomi Saito, Nathália Carolina Fernandes Fagundes, Lucianne Cople Maia, Rafael Rodrigues Lima
Frontiers in Public Health.2021;[Epub] CrossRef - Why is exercise important to dentistry?
Rishiniy Pushparatnam
BDJ Team.2021; 8(2): 20. CrossRef - The role of Wnt pathway in obesity induced inflammation and diabetes: a review
Bhabajyoti Das, Manas Das, Anuradha Kalita, Momita Rani Baro
Journal of Diabetes & Metabolic Disorders.2021; 20(2): 1871. CrossRef - Association between obesity and chronic periodontitis
Tsung-Po Chen, Hui-Chieh Yu, Tai-Hsin Lin, Yu-Hsun Wang, Yu-Chao Chang
Medicine.2021; 100(41): e27506. CrossRef - Association Between Triglyceride-Glucose Index and Risk of Periodontitis: A Cross-Sectional Study
Lili Li, Lu Li, Yi Zhou, Xu Chen, Yan Xu
International Journal of General Medicine.2021; Volume 14: 9807. CrossRef Antioxidant and Anti-Inflammatory Properties of Melatonin in Patients with Type 2 Diabetes Mellitus with Periodontal Disease Under Non-Surgical Periodontal Therapy: A Double-Blind, Placebo-Controlled Trial
Ahmad Zare Javid, Seyed Ahmad Hosseini, Hassan Gholinezhad, Leila Moradi, Mohammad Hosein Haghighi-zadeh, Hadi Bazyar
Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy.2020; Volume 13: 753. CrossRef- Influence of non-surgical periodontal therapy on insulin resistance in chronic periodontitis subjects with prediabetes
Shravani Babladi, Rupali Agnihotri, Pratibha Gopalkrishna, Shobha U. Kamath, Sushma Jogi, Brunda Arun
International Journal of Diabetes in Developing Countries.2019; 39(2): 273. CrossRef - The relationship of diabetes, periodontitis and cardiovascular disease
Anandhara Indriani Khumaedi, Dyah Purnamasari, Ika Prasetya Wijaya, Yuniarti Soeroso
Diabetes & Metabolic Syndrome: Clinical Research & Reviews.2019; 13(2): 1675. CrossRef - Association between Regular Electronic Nicotine Product Use and Self-Reported Periodontal Disease Status: Population Assessment of Tobacco and Health Survey
Nkiruka Atuegwu, Mario Perez, Cheryl Oncken, Sejal Thacker, Erin Mead, Eric Mortensen
International Journal of Environmental Research and Public Health.2019; 16(7): 1263. CrossRef - Evaluation and comparison of serum vitamin D and calcium levels in periodontally healthy, chronic gingivitis and chronic periodontitis in patients with and without diabetes mellitus – a cross-sectional study
Anshuka A. Agrawal, Abhay P. Kolte, Rajashri A. Kolte, Suresh Chari, Madhur Gupta, Resham Pakhmode
Acta Odontologica Scandinavica.2019; 77(8): 592. CrossRef - Periodontal inflammation correlates with systemic inflammation and insulin resistance in patients with recent diagnosis of type 2 diabetes
Natacha Oyarzo, María Riveros, Constanza Andaur, Jessica Liberona, Víctor Cortés
ARS MEDICA Revista de Ciencias Médicas.2019; 44(1): 6. CrossRef - A longitudinal study on the relationship between dental health and metabolic syndrome in Japan
Shin‐ichi Sakurai, Shin‐ichi Yamada, Imahito Karasawa, Akinari Sakurai, Hiroshi Kurita
Journal of Periodontology.2019; 90(7): 728. CrossRef - Triglyceride to high density lipoprotein cholesterol ratio and its association with periodontal disease in Korean adults: findings based on the 2012–2014 Korean national health and nutrition examination survey
Yu-Jin Kwon, Jeong-Won Park, Hyoung-Ji Lim, Yong-Jae Lee, Hye-Sun Lee, Jae-Yong Shim
Clinical Oral Investigations.2018; 22(1): 515. CrossRef - Periodontal complications of hyperglycemia/diabetes mellitus: Epidemiologic complexity and clinical challenge
Thomas Kocher, Jörgen König, Wenche Sylling Borgnakke, Christiane Pink, Peter Meisel
Periodontology 2000.2018; 78(1): 59. CrossRef - Nanofibrous Spongy Microspheres To Distinctly Release miRNA and Growth Factors To Enrich Regulatory T Cells and Rescue Periodontal Bone Loss
Zhongning Liu, Xin Chen, Zhanpeng Zhang, Xiaojin Zhang, Laura Saunders, Yongsheng Zhou, Peter X. Ma
ACS Nano.2018; 12(10): 9785. CrossRef - The role of dental practitioners in addressing overweight and obesity among children: A scoping review of current interventions and strategies
Amy R. Villarosa, David George, Lucie M. Ramjan, Ravi Srinivas, Ajesh George
Obesity Research & Clinical Practice.2018; 12(5): 405. CrossRef - Periodontitis and prevalence of elevated aminotransferases in the Hispanic Community Health Study/Study of Latinos
Aderonke A. Akinkugbe, A. Sidney Barritt, Jianwen Cai, Steven Offenbacher, Bharat Thyagarajan, Tasneem Khambaty, Richard Singer, Eric Kallwitz, Gerardo Heiss, Gary D. Slade
Journal of Periodontology.2018; 89(8): 949. CrossRef - Association of circulating leptin and adiponectin with periodontitis: a systematic review and meta-analysis
Junfei Zhu, Bin Guo, Xueqi Gan, Ling Zhang, Yuting He, Beilei Liu, Xin Chen, Suhan Zhang, Haiyang Yu
BMC Oral Health.2017;[Epub] CrossRef - Normal Oral Flora and the Oral Ecosystem
Lakshman Samaranayake, Victor H. Matsubara
Dental Clinics of North America.2017; 61(2): 199. CrossRef - Involvement of insulin resistance in normoglycaemic obese patients with periodontitis: A cross‐sectional study
Mayte Martinez‐Herrera, Francisco Javier Silvestre, Javier Silvestre‐Rangil, Celia Bañuls, Milagros Rocha, Antonio Hernández‐Mijares
Journal of Clinical Periodontology.2017; 44(10): 981. CrossRef - The relationship between insulin resistance and periodontitis is not affected by Mediterranean diet in a Spanish population
M. Pulido-Moran, P. Bullon, J.M. Morillo, M. Battino, J.L. Quiles, MCarmen Ramirez-Tortosa
Archives of Oral Biology.2017; 77: 62. CrossRef - Periodontitis contributes to adipose tissue inflammation through the NF-
B, JNK and ERK pathways to promote insulin resistance in a rat model
Yanli Huang, Jin Zeng, Guoqing Chen, Xudong Xie, Weihua Guo, Weidong Tian
Microbes and Infection.2016; 18(12): 804. CrossRef - Systemic Inflammatory Biomarkers and Their Association With Periodontal and Diabetes‐Related Factors in the Diabetes and Periodontal Therapy Trial, A Randomized Controlled Trial
Maria L. Geisinger, Bryan S. Michalowicz, Wei Hou, Elinor Schoenfeld, Marie Gelato, Steven P. Engebretson, Michael S. Reddy, Leslie Hyman
Journal of Periodontology.2016; 87(8): 900. CrossRef - Association of Periodontitis and Subsequent Depression
Chih-Chao Hsu, Yi-Chao Hsu, Hsuan-Ju Chen, Che-Chen Lin, Kuang-Hsi Chang, Chang-Yin Lee, Lee-Won Chong, Chia-Hung Kao
Medicine.2015; 94(51): e2347. CrossRef - Association between insulin resistance and periodontitis in Korean adults
Sang Gyu Lim, Kyungdo Han, Hyun‐Ah Kim, Sung Woon Pyo, Young‐Sik Cho, Kyung‐Soo Kim, Hyeon Woo Yim, Won‐Chul Lee, Yong Gyu Park, Yong‐Moon Park
Journal of Clinical Periodontology.2014; 41(2): 121. CrossRef - Effect of scaling and root planing combined with systemic doxycycline therapy on glycemic control in diabetes mellitus subjects with chronic generalized periodontitis: a clinical study
Subodh P. Gaikwad, Abhijit N. Gurav, Abhijeet R. Shete, Hitesh M. Desarda
Journal of Periodontal & Implant Science.2013; 43(2): 79. CrossRef - Diabetes Mellitus and Inflammation
Eric Lontchi-Yimagou, Eugene Sobngwi, Tandi E. Matsha, Andre Pascal Kengne
Current Diabetes Reports.2013; 13(3): 435. CrossRef - Bacterial Infection Increases Periodontal Bone Loss in Diabetic Rats through Enhanced Apoptosis
Sandra Pacios, Oelisoa Andriankaja, Jun Kang, Maher Alnammary, Jason Bae, Beatriz de Brito Bezerra, Helen Schreiner, Daniel H. Fine, Dana T. Graves
The American Journal of Pathology.2013; 183(6): 1928. CrossRef
 PubReader
PubReader Cite
Cite