The Role of Oxidative Stress in the Pathogenesis of Diabetic Vascular Complications
Article information
Abstract
Oxidative stress has been paid increasing attention to as an important causative factor for diabetic vascular complications. Among possible various sources, accumulating evidence has indicated that NAD(P)H oxidase may be the most important source for reactive oxygen species production in diabetic vascular tissues. The mechanisms underlying activation and up-regulation of NAD(P)H oxidase has been supposed to be mediated by high glucose-induced protein kinase C (PKC) activation. In this review article, activation of local renin-angiotensin II system induced by chymase activation is also shown to amplify such a PKC-dependent activation of NAD(P)H oxidase. Additionally, human evidence showing the beneficial effect of antioxidants on diabetic vascular complications. Bilirubin has been recognized as a strong endogenous antioxidant. Here markedly lower prevalence of vascular complications is shown in diabetic patients with Gilbert syndrome, a congenital hyperbilirubinemia, as well as reduced markers of oxidative stress and inflammation. Lastly, statin, angiotensin II receptor blocker, chymase inhibitor, bilirubin and biliverdin, PKC β isoform inhibitor, and glucagon-like peptide-1 analog, are shown to serve as antioxidants and have some beneficial effect on diabetic vascular complications, via inhibiting PKC-NAD(P)H oxidase activation, supporting the notion that this mechanism may be an effective therapeutic target for preventing diabetic vascular complications.
INTRODUCTION
It is well established that hyperglycemia is a major causative factor for micro-vascular disease. In addition, recent epidemiological reports have suggested that postprandial hyperglycemia is an important risk factor for cardiovascular diseases, although the pathological role of chronic hyperglycemia is still controversial. Several well-researched theories have been proposed to explain how chronic hyperglycemia or postprandial hyperglycemia causes the micro-and macro-vascular derangements. These theories include increased polyol pathway flux, increased advanced glycation end-product (AGE) formation, activation of protein kinase C (PKC) and increased oxidative stress. Among them, oxidative stress has been paid increasing attention to as a common mediator of these biochemical abnormalities. Among many possible sources for oxidative stress, recent reports including ours have shown that a PKC-dependent activation of NAD(P)H oxidase may be a major source for increased oxidative stress in vascular tissues of diabetes and insulin resistant state. In this article, we show that this mechanism may be an effective therapeutic target for preventing diabetic vascular complications.
INCREASED OXIDATIVE STRESS IN DIABETIC VASCULAR TISSUES AND ITS UNDERLYING MECHANISM
A number of in vitro and in vivo studies have demonstrated that many protein, lipid and DNA markers of oxidative stress are increased in vascular tissues from animals and patients with diabetes [1-4]. We also showed the increase in reactive oxygen species (ROS) production in diabetic animal models by in vivo electron spin resonance method that is non-invasive and more specific than above-mentioned oxidative stress markers [5]. Several mechanisms for increased oxidative stress have been postulated. In the process of mitochondrial respiration, several percentages of oxygen consumed is converted into superoxide. High glucose level was reported to increase such a mitochondrial superoxide production in vascular endothelial cells [6]. High glucose level also can increase ROS production through glucose auto-oxidation, increased non-enzymatic glycation of proteins and activation of xanthine oxidase. Among possible various sources, increasing evidence has indicated that NAD(P)H oxidase may be the most important source for ROS production in diabetic vascular tissues. We first reported that high glucose level stimulated ROS production in cultured vascular cells through a PKC-dependent activation of NAD(P)H oxidase [7]. The non-phagocytic NAD(P)H oxidase comprises of a membrane-associated cytochrome b558 composed of Nox family proteins (gp91phox, Nox1, and Nox4) and p22phox, and several cytosolic regulatory components, p47phox, p67phox and small GTP binding protein Rac 1 or Rac 2. Our in vitro evidence has been supported by accumulating evidence showing that the activity of NAD(P)H oxidase, in parallel with the increased level of its subunit proteins, is increased in vascular tissues form animal models and diabetic patients [5,8-11]. We also reported that the expression of essential subunits of Nox4 and p22phox in the kidney of diabetic animal models [12]. Activation of Rac-1 and translocation of p47phox and p67phox, both of which are mediated by PKC activation, are supposed to be the mechanism underlying high glucose level-induced NAD(P)H oxidase activation [13,14]. The superoxide production from NAD(P)H oxidase is controlled by its expression dynamics as well as by its activation. The mechanism for increased expression of NAD(P)H oxidase subunits in diabetic vascular tissues remained to be well established, since the regulatory mechanism of the expression of NAD(P)Oxidase subunits is generally not fully understood. In the knockout mouse of the PKC β isoform, the normalization of increased expression of Nox4, oxidative stress, proteinuria and tissue abnormalities in diabetic kidney was observed [15]. These findings also support the role of PKC activation in the increased expression of NAD(P)H oxidase subunits. The detailed molecular mechanism should be clarified in the further studies.
ROLE OF LOCAL RENIN-ANGIOTENSIN SYSTEM IN OXIDATIVE STRESS
Experimental and clinical trials have suggested that blockade of the renin-angiotensin system (RAS) with angiotensin 1-converting enzyme (ACE) inhibitors or angiotensin II (ATII) receptor blocker (ARB) have some protective effects on diabetic nephropathy and cardiovascular events, which appear to be independent of its antihypertensive effect. ATII was reported to mediate NAD(P)H oxidase-dependent superoxide production via PKC activation. We reported the improvement of Nox4 expression and oxidative stress in the kidney of diabetic mice by administration of ARB [16]. ACE is involved in the production of tissue ATII, but it is recently suggested that chymase has more efficient tissue ATII-forming activities than ACE in several species including human and hamsters. In addition, several reports have shown that chymase expression may be up-regulated in diabetic renal tissues [17]. We have confirmed that chymase expression increases in renal tissues, aorta and cardiac tissues of streptozotocin-induced diabetic hamster and examined the effect of chymase-specific inhibitor [18]. Oral administration of the chymase-specific inhibitors protected against elevated intra-renal ATII concentrations, Nox4 expression, oxidative stress, and renal dysfunction including proteinuria and mesangial expansion in streptozotocin-induced diabetic hamsters [18]. It also normalized oxidative stress, Nox4 expression and myocardial fibrosis in diabetic hamsters [19]. Because low-grade inflammation is known to occur in diabetic vascular tissues, including renal tissues, infiltrated inflammatory cells, such as mast cells, may be the source of the increased chymase levels. High glucose is the initiator of increased oxidative stress and inflammation, but the subsequent increases in chymase expression and local ATII production may further accelerate ROS production and inflammation, ultimately forming a vicious cycle of oxidative stress and inflammation, although this hypothesis should be evaluated in future studies.
HUMAN EVIDENCE FOR THE EFFECTIVENESS OF ANTIOXIDANT ON DIABETIC COMPLICATIONS
Although the involvement of oxidative stress in diabetic vascular complications has been demonstrated in vitro and in animal studies, the effectiveness of antioxidant therapy is still controversial because there have been very few human studies indicating the beneficial effects of antioxidants on diabetic vascular complications. Bilirubin has been recognized as an important endogenous antioxidant [20] and also reported to have an inhibitory effect on the activity of NAD(P)H oxidase [21]. We therefore compared the prevalence of vascular complications in patients with diabetes and Gilbert syndrome (a congenital hyperbilirubinemia) and in patients with diabetes without Gilbert syndrome [22]. The adjusted odds ratio for the association of Gilbert syndrome with retinopathy was 0.22 (P<001); with macroalbuminuria, 0.20 (P=0.01); with coronary artery disease, 0.21 (P=0.04); and with cerebrovascular disease, 0.50 (P=0.22) (Table 1). This study showed for the first time the lower prevalence of vascular complications in patients with diabetes and Gilbert syndrome, as well as reduced markers of oxidative stress and inflammation (Fig. 1). These findings strongly suggest that antioxidant therapy may be useful for preventing the development of diabetic vascular complications. Furthermore, serum bilirubin concentrations were demonstrated to be negatively associated with albuminuria in patients with type 2 diabetes [23]. Another group of Japanese investigators reported in their cross-sectional study on more than 3,000 participants, that compared to subjects with in the lowest bilirubin quartile, those with the highest bilirubin levels had a four times lower prevalence of diabetic retinopathy [24]. Interestingly, our cross-sectional study of 12,949 general persons revealed an inverse relation between serum bilirubin level and the prevalence of type 2 diabetes [25]. In a large Korean cross-sectional study on almost 94,000 subjects, high serum bilirubin was also found to be associated with the reduced risk of diabetes mellitus and diabetic nephropathy [26]. Consistent with these data, low prevalence of metabolic syndrome in subjects with phenotypic Gilbert syndrome was described in a recent large Korean study on more than12,000 participants [27]. Antioxidant therapy may be useful for inhibiting the onset of diabetes itself as well as inhibiting diabetic complications.
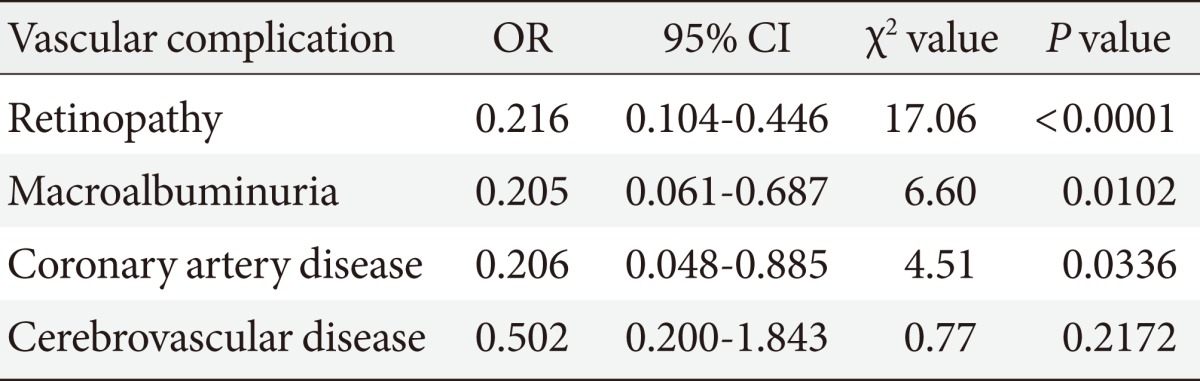
Adjusted odds ratio for retinopathy, macroalbuminuria, coronary artery disease and cerebrovascular disease in diabetic patients with Gilbert syndrome compared with those without it
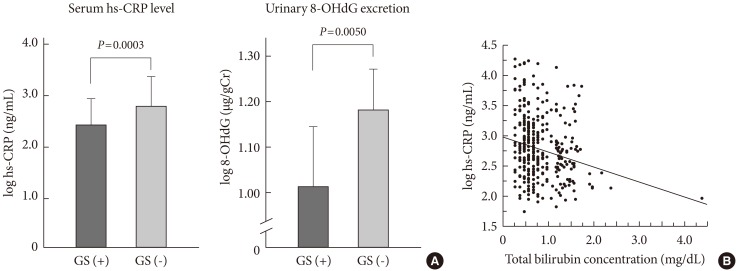
Serum high sensitivity C-reactive protein (hs-CRP) levels and the amounts of urinary 8-hydroxy-2'-deoxyguanosine (8-OHdG) excretion. (A) Comparison between diabetic patients with Gilbert syndrome, GS (+), and those without it, GS (-). Due to their skewed distribution, serum hs-CRP levels (ng/mL) and the amounts of urinary 8-OHdG excretion (urinary 8-OHdG to creatinine ratio, µg/gCr) were log10 transformed before all analyses. (B) Correlation between total bilirubin levels and serum hs-CRP levels. Spearman correlation analysis: r=-0.213, P<0.0001.
ANTIOXIDANT THERAPY TARGETING THE MECHANISM FOR PKC-NAD(P)H OXIDASE ACTIVATION
We have proposed an antioxidant therapy using the drugs or chemicals which target the mechanism for PKC-NAD(P)H oxidase activation for preventing diabetic vascular complications [28]. The possible mechanism underlying PKC-NAD(P)H oxidase activation and drugs or chemicals targeting its mechanism are shown in Fig. 2.

The mechanism underlying increased oxidative stress in diabetic vascular tissues and possible antioxidant therapies targeting its mechanism for inhibiting diabetic vascular complications. ARB, angiotensin II receptor blocker; PKC, protein kinase C; ATII, angiotensin II; GLP-1, glucagon-like peptide-1.
Statin
We noted the ability of statin to inhibit geranylgeranylation of small GTP binding protein Rho family including Rac. We found that pitavastatin inhibited high glucose-induced increase in ROS production in parallel with inhibition of Rac-1 activity in cultured aortic smooth muscle cells [13]. Such inhibition by pitavastatin was reversed by coincubation with mevalonic acid. In addition, we found that pitavastatin treatment prevented the increased free ROS production in diabetic rats using the above-mentioned in vivo electron spin resonance method [13]. Furthermore, we showed that pitavastatin administration improved albuminuria and histologic abnormalities such as increased renal mesangial expansion in db/db mice, along with the improvement of renal Nox4 expression and oxidative stress [29]. The results in recent our clinical trial may be in support for the effectiveness of pitavastatin treatment on albuminuria in diabetic patients [30].
ARB and chymase-specific inhibitor
In addition to the beneficial effect on vascular complications, we have shown that oral administration of ARB attenuated the increased expression of NAD(P)H oxidase in pancreatic islets together with the inhibition of oxidative stress and restored decreased insulin contents in islets of db/db mice [31]. These drugs may have a protective effect on the onset of type 2 diabetes.
Bilirubin and biliverdin
To directly examine the beneficial effect of bilirubin and biliverdin (a precursor of bilirubin), we determined whether hereditary hyperbilirubinemic Gunn rats and biliverdin-treated diabetic db/db mice were resistant to the development of renal dysfunction. Both rodent models had less albuminuria and complete protection against the progression of mesangial expansion, accompanied by the normalization of oxidative stress and Nox4 expression [32]. These results suggested that biliverdin and biliverdin may protect against diabetic nephropathy and this may lead to a novel antioxidant therapy for diabetic nephropathy. Similar beneficial effect was also found in oral administration of phycocyanin derived from Spirulina platensis, a blue-green algae, and its chromophore phycocyanobilin, which has a chemical structure similar to that of biliverdin (unpublished data).
In addition, biliverdin treatment delayed progressive worsening of glucose tolerance in db/db mice mainly via inhibition of oxidative stress-induced β-cell damage, suggesting the possible effectiveness of antioxidant therapy for preventing the onset type 2 diabetes as well as preventing diabetic complications [33].
PKC β2 inhibitor
Among various isoforms, preferential activation of PKC β isoform is reported to occur in many vascular tissues in the diabetic state [34]. PKC β isoform-specific inhibitor LY33531 improves dysfunction and tissue changes in kidney and retina of the experimental diabetic animal model [35] and the results in the clinical trial are reported to be useful [36], although it does not yet reach a clinical application. The participation of the antioxidant action is presumed in part of the mechanism.
Glucagon-like peptide-1 (GLP-1) analog
Currently, the GLP-1 receptor agonist exendin-4 and the GLP-1 analog liraglutide are used to treat type 2 diabetes. The major downstream pathway of GLP-1 receptor activation is generation of the second messenger cAMP followed by activation of PKA or Epac2, and NAD(P)H oxidases have been reported to be inhibited by PKA activation in phagocytes. We therefore hypothesized that GLP-1 receptor agonists may inhibit renal NAD(P)H oxidase. In the streptozotocin-induced type 1 diabetes models, liraglutide treatment did not significantly affect plasma glucose levels or body weights, but it still normalized urinary albuminuria in diabetic rats in parallel with normalization of oxidative stress and expression of renal NAD(P)H oxidase components (Nox4, gp91phox, p22phox, p47phox), independently of a glucose-lowering effect [37]. Liraglutide inhibited NADPH-dependent superoxide production from cultured renal mesangial cells, and its inhibitory effect was reversed by the adenylate cyclase inhibitor SQ22536 and the PKA inhibitor H89, but not reversed by Epac2 inhibition. These results suggested that the effect of liraglutide on NAD(P)H oxidase was mediated by the cAMP-PKA pathway. Its clinical efficacy should be tested in human trial.
CONCLUSION
In this review, the experimental and clinical studies on the role of oxidative stress in the development of diabetic vascular complication and the molecular mechanism underlying oxidative stress are discussed. These studies strongly show that antioxidant therapy is potentially effective to protect against diabetic vascular complications and one of the therapeutic targeting mechanism is activation or up-regulation of PKC-NAD(P)H oxidase. In this article, the effectiveness of several drugs or chemicals which can inhibit this mechanism is shown. These drug or chemicals are expected to be clinically effective for preventing diabetic vascular complications, but the development of more effective drugs for clinical use is also expected.
ACKNOWLEDGMENTS
We appreciate the excellent assistance of Ms. Eri Nagashima for making this manuscript. This work was supported in part by the Special Coordination Funds for Promoting Science and Technology, Japan (SCF funding program "Innovation Center for Medical Redox Navigation").
Notes
No potential conflict of interest relevant to this article was reported.