- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 38(2); 2014 > Article
-
ReviewPathophysiology Altered Transendothelial Transport of Hormones as a Contributor to Diabetes
- Nanyoung Yoon, Thanh Q. Dang, Helen Chasiotis, Scott P. Kelly, Gary Sweeney
-
Diabetes & Metabolism Journal 2014;38(2):92-99.
DOI: https://doi.org/10.4093/dmj.2014.38.2.92
Published online: April 18, 2014
Department of Biology, York University, Toronto, ON, Canada.
- Corresponding author: Gary Sweeney. Department of Biology, York University, 4700 Keele Street, Toronto, ON, M3J 1P3, Canada. gsweeney@yorku.ca
Copyright © 2014 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The vascular endothelium is a dynamic structure responsible for the separation and regulated movement of biological material between circulation and interstitial fluid. Hormones and nutrients can move across the endothelium either via a transcellular or paracellular route. Transcellular endothelial transport is well understood and broadly acknowledged to play an important role in the normal and abnormal physiology of endothelial function. However, less is known about the role of the paracellular route. Although the concept of endothelial dysfunction in diabetes is now widely accepted, we suggest that alterations in paracellular transport should be studied in greater detail and incorporated into this model. In this review we provide an overview of endothelial paracellular permeability and discuss its potential importance in contributing to the development of diabetes and associated complications. Accordingly, we also contend that if better understood, altered endothelial paracellular permeability could be considered as a potential therapeutic target for diabetes.
- Structure and function of endothelium
- A monolayer of endothelial cells lines the entire circulatory system of the body. This endothelium acts as a barrier which regulates the exchange of hormones, proteins and small molecules between the vascular compartment and the interstitial space [1,2]. The actions of a hormone or nutrient on a target tissue is implicitly dependent upon the ability of these factors to gain access to the target and numerous studies have indicated that hormone and nutrient concentrations in blood differ from those surrounding cells on the tissue side of the blood vessel endothelium [3,4,5,6,7,8,9,10,11]. In this regard, it is our contention that the significance of the endothelium as a regulator of hormone and substrate access to target tissues is often underappreciated. Endothelial permeability can be regulated by two distinct pathways: 1) the transcellular pathway where solutes are actively transported across the endothelium, primarily via caveolae-mediated transcytosis; or 2) the paracellular pathway where solutes passively move through the intercellular space between adjacent endothelial cells (Fig. 1).
INTRODUCTION
- The transcellular movement of solutes involves energy-dependent trafficking of vesicles across the endothelium [2]. This often requires their recognition by receptors in caveolae on the luminal surface of the endothelium, may involve vesiculo-vacuolar organelles or occur via transcellular channels [2]. Caveolae, cholesterol- and sphingolipid-rich non-clathrin-coated pits, are abundant in endothelial cells. After ligand binding to receptors in caveolae, dynamin-mediated endocytosis occurs followed by vectorial transport and fusion of the vesicle with basolateral membrane, resulting in the release of contents by exocytosis [2]. As a general concept, transcellular vesicle trafficking is important in the transport of larger macromolecules across the endothelium since the paracellular route is typically capable of restricting passage of solutes larger than 3 nm in radius [12,13].
TRANSCELLULAR TRANSPORT
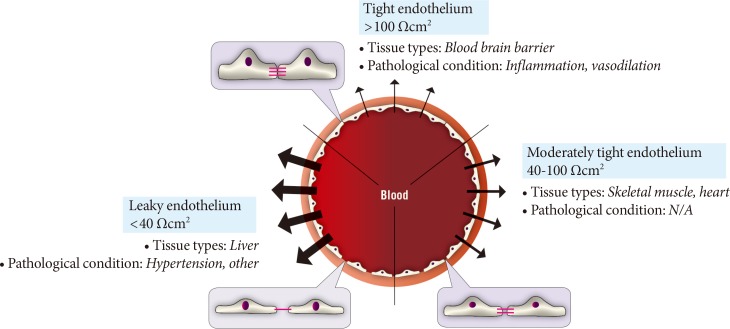
- The paracellular mode of transport across endothelia depends upon concentration gradients between blood and interstitial fluid. One regulatory mechanism of paracellular movement involves tight junctions (TJs) which are composed of strands of transmembrane and cytosolic proteins that closely control paracellular flux and have been established to be important in regulating hormone transport [4,12,14]. As a component of the cell-cell junction of endothelia, the TJ provides a selective barrier to solute movement between cells [13]. The permselectivity of the TJ barrier is dependent on the variable assemblage of TJ proteins that comprise the complex. Proteins such as occludin, tricellulin, and claudins directly establish the TJ barrier and form the backbone of TJ strands while the cytosolic proteins, such as ZO-1, provide structural support to the TJ complex. The incorporation of specific TJ protein isoforms can enhance TJ barrier function (i.e., make TJs tighter) or form channels to increase TJ permeability (i.e., make TJs leakier) [13]. Importantly, skeletal muscle and heart vasculature have continuous endothelium with TJs between cells (Fig. 2) [15,16]. However, the liver and spleen have a discontinuous endothelium with large holes, which allow rapid equilibration of plasma with the underlying tissue [15,16]. It was suggested that this expedited exposure of the liver to plasma solute changes may partly explain its earlier susceptibility to insulin resistance [17]. Indeed, the precise architecture of TJs varies between different vascular beds, with TJs being either dominant at the apical point of intercellular space or intermingled with adherens junctions (AJs). Therefore, there is no doubt that transendothelial paracellular flux in certain vascular beds is more susceptible to changes in TJ composition and structure. Furthermore, changes in paracellular transport leading to endothelial hyperpermeability is a significant problem in vascular inflammation associated with diabetes, cancer, ischaemia-reperfusion injury, thrombisis, trauma, sepsis, and adult respiratory distress syndrome [18]. Importantly, despite the tremendous interest in adipokines over recent years very few studies have examined the critical step whereby and factors secreted by adipose tissue, or other tissues, must enter the circulation via capillaries or lymph. Paracellular movement is very likely to be a major regulatory step in this process. Indeed, it has been demonstrated that lymph/capillary partitioning of adipokines is dependent upon their molecular size and proposed that this will have important ramifications for their regional and systemic distribution and subsequent physiological effects [19].
- It is also important to realize that crosstalk exists between 1) transcellular and paracellular routes of transport across endothelia and 2) TJs and AJs of endothelia. An example of the former is that in caveolin-1 knockout mice, or upon siRNA-mediated reduction of caveolin-1, altered TJ assembly in small capillaries and veins increased paracellular transport of albumin [20,21,22]. In the latter case, it has been shown that vascular endothelial (VE)-cadherin mediated signaling can regulate the expression of TJ proteins and consequently alter barrier function [2].
- Indeed, evidence suggests that AJs also play a crucial role in regulating vascular permeability. They are comprised of calcium-dependent VE-cadherin in complex with a range of interacting partners, such as β-catenin and p120-catenin [2] and the expression of VE-cadherin is a specific early developmental marker for endothelial lineage. Deletion of the VE-cadherin gene induces embryonic lethality due to early collapse of the vasculature [23]. Increased vascular permeability occurs when VE-cadherin homophilic binding is inhibited. AJ stability is also critically regulated by phosphorylation [2]. More specifically, phosphorylation of VE-cadherin at catenin binding sites induced internalisation and thus alterations in vascular permeability. Interestingly, recent work demonstrated that phosphorylation of these residues (tyrosine 658 and 685 of VE-cadherin) occurs constitutively in veins but not arteries, correlating with enhanced leakiness in venous vessels [24]. Furthermore, single point mutations in VE-cadherin (Y658F and Y685F) reduce paracellular permeability by blocking internalisation of VE-cadherin [24].
PARACELLULAR TRANSPORT
- The concept of endothelial dysfunction in diabetes is well established [25]. However, this typically refers to vascular functions independent of transendothelial solute flux. The endothelium is also a dynamic interface that responds to various stimuli and synthesizes and liberates vasoactive molecules such as nitric oxide, prostaglandins and endothelin. Accordingly, vascular complications in diabetes include outcomes occurring in large (atherosclerosis, cardiomyopathy) and small (retinopathy, nephropathy, neuropathy) vessels [26]. We believe that diabetes-induced deleterious alterations in paracellular solute movement also represents a form of endothelial dysfunction which should be fully characterized to establish its physiological significance.
ENDOTHELIAL DYSFUNCTION IN DIABETES
- Transport of insulin has been perhaps the best studied example. Insulin concentration at the target cell surface has been shown to be very different from that in plasma by approaches including microdialysis [7,8], direct interstitial sampling [9], and lymph measurements [5,6]. Furthermore, early temporal studies have demonstrated that insulin-mediated glucose uptake in muscle lags behind increases in plasma insulin [27], In contrast, when using cultured skeletal muscle cells, the addition of recombinant insulin has been shown to stimulate glucose uptake almost immediately (maximal at~10 minutes) [28]. The time delay seen in vivo has led to the suggestion that reaching the interstitial space is the limiting factor for insulin-mediated glucose uptake [14]. Thus, modification of access to skeletal muscle can have major effects on insulin action and subsequent metabolism [14,29]. Indeed, the efficiency and extent of insulin delivery to the interstitial space can be inhibited physiologically by diet [30]. The mechanism via which insulin crosses the endothelium is at least in part via the paracellular pathway and for more information on this topic readers are referred to recent excellent review articles by Kolka and Bergman [14,31].
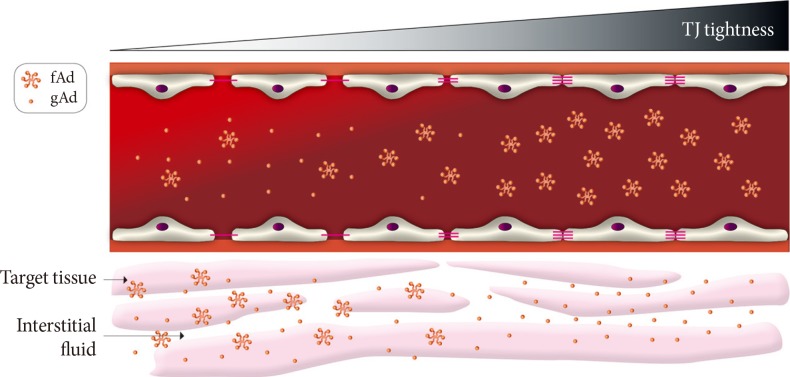
- Recent years have seen great interest in the role of adiponectin in the pathophysiology of diabetic complications [32,33,34]. Numerous studies have now established that adiponectin can act on various targets to mediate antidiabetic, anti-inflammatory, antiatherosclerotic, and cardioprotective effects [32,34,35]. Thus, enhancing adiponectin action is considered as a therapeutically beneficial strategy. Adiponectin circulates in the blood as three multimeric complexes; low molecular weight (LMW; trimer), medium molecular weight (MMW; hexamer) and high molecular weight (HMW; oligomer), and in obese and diabetic patients reduced circulating levels of adiponectin have been observed. In addition, cleavage of adiponectin by leukocyte derived elastase generates a C-terminal globular fragment of adiponectin which mediates important functional effects. It is likely that delivery of adiponectin to the interstitial space is a major, yet underestimated, determinant of its function. Given the size range of the biologically active forms of adiponectin (16 kDa for globular form and >500 kDa for HMW) there is considerable potential for selective paracellular transport of adiponectin, particularly in different vascular beds. HMW adiponectin is often considered to be the most biologically active and most relevant form with respect to the metabolic syndrome, and we speculate that given the large size of the HMW form, transendothelial movement may prove to be a significant rate-limiting step in its physiological actions (Fig. 3). Indeed, it has been proposed that HMW adiponectin mediates beneficial metabolic effects primarily by acting on liver [34], whereas the globular form is thought to have more potent metabolic effects in skeletal muscle [33]. These observations could be explained by the leaky and moderately tight paracellular barriers present in vascular beds of these respective tissues, although this is likely an oversimplification (Fig. 3). As far as we are aware, no studies have directly examined the paracellular movement of adiponectin in skeletal muscle and liver.
- Studies of adiponectin movement across the blood-brain barrier (BBB) have indicated that only LMW and MMW, but not HMW forms were found in cerebralspinal fluid [36,37,38]. Surprisingly, there are only a few studies examining interstitial adiponectin levels. One found that interstitial concentrations of adiponectin in human adipose tissue were ~25-fold lower than plasma [39] while the other found that exercise increased interstitial adiponectin levels, which were around 20% of plasma concentration [40]. We hypothesize that the magnitude of gradient between plasma and interstitial levels of adiponectin in skeletal muscle may be higher due to existence of a tighter endothelial barrier within the muscle vasculature. Indeed, this may be highly significant as it has been shown that hyperinsulinemia differentially affects the compartmental (interstitial and circulating) distribution of the adiponectin complexes in lean and insulin-resistant, obese individuals [41]. We have recently shown that adiponectin moves across cultured endothelial monolayers via a paracellular route and that this movement was reduced by hormonal or physical manipulation of TJs.
STUDIES ON TRANSENDOTHELIAL TRANSPORT OF HORMONES
- Studies examining changes in TJ composition and protein expression in diabetes have not been extensive but have so far yielded numerous consistent observations in the study of diabetic complications. For example, reductions in ZO-1 have been observed in kidney glomeruli, the retina, BBB and intestine of various diabetic models [42,43,44,45,46]. Localization of ZO-1 in glomeruli was also investigated by electron microscopy and found to redistribute from the podocyte membrane to the cytoplasm in the diabetic kidney [47]. Several reports have indicated that both occludin and claudin-5 were reduced in diabetic retina or BBB [44,45,46,48,49]. Hence, expression or localization of TJ proteins appear to be susceptible to a diabetic environment and contribute to complications such as retinopathy and nephropathy. More widespread analysis is now needed, particularly in metabolically active tissues. Knockout mouse models have been generated for various TJ proteins, including occludin and claudin-1, 2, 4, 5, 7, 9, 11, 14, 15, 16, 18, and 19 [50,51,52,53,54,55,56,57,58,59,60,61,62,63]. Little to no information was given regarding their metabolic phenotype, however in one study using Cldn2-/-Cldn15-/- double-knockout mice the animals died from malnutrition related to defective absorption of glucose, amino acids and fats [63]. These models may prove valuable in elucidating the significance of alterations in transendothelial hormone flux in determining metabolic dysfunction in diabetes.
TJ COMPOSITION AND PROTEIN EXPRESSION IN DIABETES
- Based on the above description of the importance of endothelial transport in diabetes, it is evident that developing therapeutic strategies which manipulate paracellular flux may prove useful. Indeed, a literature review indicates that various agents are already available; such as peptides which bind to integral TJ proteins, siRNA and antisense oligonucleotides targeting TJ proteins, various toxins, lipids, and activators or inhibitors of kinases and phosphatases that regulate junction assembly and function have all been shown to regulate paracellular permeability [64]. Nevertheless, there is an innate risk in therapeutics which globally alter transendothelial permeability and so it will be desirable to elicit changes in a localized or tissue-specific manner, or to targeting specific components of TJs which allow more controlled and selective changes in permeability [65]. One example of a current therapeutic approach targeting TJs is the use of glucocorticoids as a locally applied treatment for diabetic retinopathy. The mechanism of action is thought to involve, at least in part, restoration of barrier function in the retinal vasculature by modifying TJ composition and structure [66]. Therefore, it is attractive to speculate that controlled manipulation of paracellular transport may be applicable to improving metabolic dysfunction in diabetes.
THERAPEUTIC POTENTIAL OF TARGETING ENDOTHELIAL TRANSPORT
- The endothelial monolayer is an important rate-limiting determinant of hormone and substrate access to target tissues. Previous studies focusing on insulin suggest that impaired delivery of insulin from plasma to interstitial space in obesity may contribute to metabolic insulin resistance and diabetes. Since adiponectin is known to mediate beneficial antidiabetic effects, and the molecular weight of biologically active forms of adiponectin ranges widely, we propose that paracellular transport may be a major determinant of transendothelial adiponectin flux. In particular, the research community has often accepted that fact that the HMW oligomeric form of adiponectin targets liver and not muscle whereas the small globular portion has 'more potency' in skeletal muscle. This was initially attributed to the higher binding affinity of globular fragment of adiponectin to skeletal muscle cell membranes which were thought to express more adiponectin receptor AdipoR1 isoform. However, such data are not consistent and easier access of the smaller globular adiponectin across the relatively tight endothelial monolayer found in skeletal muscle vasculature, in comparison with liver, may be significant. Thus, TJ-mediated, or AJ-mediated, alterations in adiponectin access to tissues from the vasculature are likely to be of important functional significance. Numerous studies in kidney and retina have indicated that expression of various TJ proteins is altered in diabetes; however, the changes in metabolic target tissues of adiponectin and their physiological significance still need to be established. The principal mechanisms responsible for altered TJ composition and structure in diabetes must also be elucidated, with likely mediators including hyperglycemia, hyperinsulinemia, free fatty acids or tumor necrosis factor-α.
- Overall, the paracellular permeability of the endothelial barrier is a tightly regulated process which can be dynamically adjusted in response to various mediators. The potential contribution to metabolic dysfunction in diabetes is apparent yet the precise significance has likely been somewhat underestimated. We believe that further investigation of TJ-mediated changes in hormone or substrate flux in diabetes and characterization of their functional significance will shed additional light on the pathophysiology of disease and identify potential therapeutic opportunities.
CONCLUSIONS
-
Acknowledgements
- Related work in the authors laboratories is funded by Canadian Diabetes Association (G.S.), Heart & Stroke Foundation of Canada (G.S.), Canadian Institutes of Health Research (G.S.), and Natural Sciences and Engineering Research Council (S.P.K.). We also wish to express sincere thanks to Haemin Kwak, School of the Art Institute of Chicago (haeminkwak88@gmail.com) for graphic design expertise in producing all figures in this manuscript.
ACKNOWLEDGMENTS
- 1. Pries AR, Kuebler WM. Normal endothelium. Handb Exp Pharmacol 2006;(176 Pt 1):1-40. Article
- 2. Goddard LM, Iruela-Arispe ML. Cellular and molecular regulation of vascular permeability. Thromb Haemost 2013;109:407-415. ArticlePubMedPMC
- 3. Chiu JD, Kolka CM, Richey JM, Harrison LN, Zuniga E, Kirkman EL, Bergman RN. Experimental hyperlipidemia dramatically reduces access of insulin to canine skeletal muscle. Obesity (Silver Spring) 2009;17:1486-1492. ArticlePubMedPMCPDF
- 4. Kolka CM, Harrison LN, Lottati M, Chiu JD, Kirkman EL, Bergman RN. Diet-induced obesity prevents interstitial dispersion of insulin in skeletal muscle. Diabetes 2010;59:619-626. ArticlePubMedPDF
- 5. Chiu JD, Richey JM, Harrison LN, Zuniga E, Kolka CM, Kirkman E, Ellmerer M, Bergman RN. Direct administration of insulin into skeletal muscle reveals that the transport of insulin across the capillary endothelium limits the time course of insulin to activate glucose disposal. Diabetes 2008;57:828-835. ArticlePubMedPDF
- 6. Yang YJ, Hope ID, Ader M, Bergman RN. Importance of transcapillary insulin transport to dynamics of insulin action after intravenous glucose. Am J Physiol 1994;266:E17-E25. ArticlePubMed
- 7. Sjostrand M, Holmang A, Lonnroth P. Measurement of interstitial insulin in human muscle. Am J Physiol 1999;276:E151-E154. ArticlePubMed
- 8. Herkner H, Klein N, Joukhadar C, Lackner E, Langenberger H, Frossard M, Bieglmayer C, Wagner O, Roden M, Muller M. Transcapillary insulin transfer in human skeletal muscle. Eur J Clin Invest 2003;33:141-146. ArticlePubMedPDF
- 9. Bodenlenz M, Schaupp LA, Druml T, Sommer R, Wutte A, Schaller HC, Sinner F, Wach P, Pieber TR. Measurement of interstitial insulin in human adipose and muscle tissue under moderate hyperinsulinemia by means of direct interstitial access. Am J Physiol Endocrinol Metab 2005;289:E296-E300. ArticlePubMed
- 10. Maggs DG, Jacob R, Rife F, Lange R, Leone P, During MJ, Tamborlane WV, Sherwin RS. Interstitial fluid concentrations of glycerol, glucose, and amino acids in human quadricep muscle and adipose tissue: evidence for significant lipolysis in skeletal muscle. J Clin Invest 1995;96:370-377. ArticlePubMedPMC
- 11. Barrett EJ, Wang H, Upchurch CT, Liu Z. Insulin regulates its own delivery to skeletal muscle by feed-forward actions on the vasculature. Am J Physiol Endocrinol Metab 2011;301:E252-E263. ArticlePubMedPMC
- 12. Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev 2013;93:525-569. ArticlePubMedPMC
- 13. Gunzel D, Fromm M. Claudins and other tight junction proteins. Compr Physiol 2012;2:1819-1852. PubMed
- 14. Kolka CM, Bergman RN. The barrier within: endothelial transport of hormones. Physiology (Bethesda) 2012;27:237-247. ArticlePubMedPMC
- 15. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 2007;100:158-173. PubMed
- 16. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 2007;100:174-190. PubMed
- 17. Kim SP, Ellmerer M, Van Citters GW, Bergman RN. Primacy of hepatic insulin resistance in the development of the metabolic syndrome induced by an isocaloric moderate-fat diet in the dog. Diabetes 2003;52:2453-2460. ArticlePubMedPDF
- 18. Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med 2009;11:e19ArticlePubMedPMC
- 19. Miller NE, Michel CC, Nanjee MN, Olszewski WL, Miller IP, Hazell M, Olivecrona G, Sutton P, Humphreys SM, Frayn KN. Secretion of adipokines by human adipose tissue in vivo: partitioning between capillary and lymphatic transport. Am J Physiol Endocrinol Metab 2011;301:E659-E667. ArticlePubMed
- 20. Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP. Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice: treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 2002;277:40091-40098. PubMed
- 21. Miyawaki-Shimizu K, Predescu D, Shimizu J, Broman M, Predescu S, Malik AB. siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am J Physiol Lung Cell Mol Physiol 2006;290:L405-L413. ArticlePubMed
- 22. Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 2001;276:38121-38138. ArticlePubMed
- 23. Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, Huber P. Role of vascular endothelial-cadherin in vascular morphogenesis. Development 1999;126:2093-2102. ArticlePubMedPDF
- 24. Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, Kurtcuoglu V, Poulikakos D, Baluk P, McDonald D, Grazia Lampugnani M, Dejana E. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 2012;3:1208ArticlePubMedPMCPDF
- 25. Ding H, Triggle CR. Endothelial dysfunction in diabetes: multiple targets for treatment. Pflugers Arch 2010;459:977-994. ArticlePubMedPDF
- 26. Symons JD, Abel ED. Lipotoxicity contributes to endothelial dysfunction: a focus on the contribution from ceramide. Rev Endocr Metab Disord 2013;14:59-68. ArticlePubMedPMCPDF
- 27. Freidenberg GR, Suter S, Henry RR, Nolan J, Reichart D, Olefsky JM. Delayed onset of insulin activation of the insulin receptor kinase in vivo in human skeletal muscle. Diabetes 1994;43:118-126. ArticlePubMed
- 28. Somwar R, Kim DY, Sweeney G, Huang C, Niu W, Lador C, Ramlal T, Klip A. GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem J 2001;359:639-649. ArticlePubMedPMCPDF
- 29. Barrett EJ, Rattigan S. Muscle perfusion: its measurement and role in metabolic regulation. Diabetes 2012;61:2661-2668. PubMedPMC
- 30. Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, Inoue M, Itoh S, Takamoto I, Sasako T, Kumagai K, Kawai T, Hashimoto S, Kobayashi T, Sato M, Tokuyama K, Nishimura S, Tsunoda M, Ide T, Murakami K, Yamazaki T, Ezaki O, Kawamura K, Masuda H, Moroi M, Sugi K, Oike Y, Shimokawa H, Yanagihara N, Tsutsui M, Terauchi Y, Tobe K, Nagai R, Kamata K, Inoue K, Kodama T, Ueki K, Kadowaki T. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab 2011;13:294-307. ArticlePubMed
- 31. Kolka CM, Bergman RN. The endothelium in diabetes: its role in insulin access and diabetic complications. Rev Endocr Metab Disord 2013;14:13-19. ArticlePubMedPMCPDF
- 32. Scheid MP, Sweeney G. The role of adiponectin signaling in metabolic syndrome and cancer. Rev Endocr Metab Disord Epub 2013 Sep 10. http://dx.doi.org/10.1007/s11154-013-9265-5.ArticlePDF
- 33. Liu Y, Sweeney G. Adiponectin action in skeletal muscle. Best Pract Res Clin Endocrinol Metab 2014;28:33-41. ArticlePubMed
- 34. Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab 2013;2:133-141. ArticlePubMedPMC
- 35. Park M, Sweeney G. Direct effects of adipokines on the heart: focus on adiponectin. Heart Fail Rev 2013;18:631-644. ArticlePubMedPDF
- 36. Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, Ezaki O, Nagai R, Tobe K, Terauchi Y, Ueki K, Minokoshi Y, Kadowaki T. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab 2007;6:55-68. ArticlePubMed
- 37. Neumeier M, Weigert J, Buettner R, Wanninger J, Schaffler A, Muller AM, Killian S, Sauerbruch S, Schlachetzki F, Steinbrecher A, Aslanidis C, Scholmerich J, Buechler C. Detection of adiponectin in cerebrospinal fluid in humans. Am J Physiol Endocrinol Metab 2007;293:E965-E969. ArticlePubMed
- 38. Kusminski CM, McTernan PG, Schraw T, Kos K, O'Hare JP, Ahima R, Kumar S, Scherer PE. Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum. Diabetologia 2007;50:634-642. ArticlePubMedPDF
- 39. Nielsen NB, Hojbjerre L, Sonne MP, Alibegovic AC, Vaag A, Dela F, Stallknecht B. Interstitial concentrations of adipokines in subcutaneous abdominal and femoral adipose tissue. Regul Pept 2009;155:39-45. ArticlePubMed
- 40. Hojbjerre L, Rosenzweig M, Dela F, Bruun JM, Stallknecht B. Acute exercise increases adipose tissue interstitial adiponectin concentration in healthy overweight and lean subjects. Eur J Endocrinol 2007;157:613-623. ArticlePubMed
- 41. Murdolo G, Hammarstedt A, Schmelz M, Jansson PA, Smith U. Acute hyperinsulinemia differentially regulates interstitial and circulating adiponectin oligomeric pattern in lean and insulin-resistant, obese individuals. J Clin Endocrinol Metab 2009;94:4508-4516. ArticlePubMed
- 42. Omran OM. Effects of thymoquinone on STZ-induced diabetic nephropathy: an immunohistochemical study. Ultrastruct Pathol 2014;38:26-33. ArticlePubMed
- 43. Shan CY, Yang JH, Kong Y, Wang XY, Zheng MY, Xu YG, Wang Y, Ren HZ, Chang BC, Chen LM. Alteration of the intestinal barrier and GLP2 secretion in Berberine-treated type 2 diabetic rats. J Endocrinol 2013;218:255-262. ArticlePubMed
- 44. Shin JY, Sohn J, Park KH. Chlorogenic acid decreases retinal vascular hyperpermeability in diabetic rat model. J Korean Med Sci 2013;28:608-613. ArticlePubMedPMCPDF
- 45. Bhattacharjee PS, Huq TS, Potter V, Young A, Davenport IR, Graves R, Mandal TK, Clement C, McFerrin HE, Muniruzzaman S, Ireland SK, Hill JM. High-glucose-induced endothelial cell injury is inhibited by a Peptide derived from human apolipoprotein E. PLoS One 2012;7:e52152ArticlePubMedPMC
- 46. Liu C, Wu J, Zou MH. Activation of AMP-activated protein kinase alleviates high-glucose-induced dysfunction of brain microvascular endothelial cell tight-junction dynamics. Free Radic Biol Med 2012;53:1213-1221. ArticlePubMedPMC
- 47. Rincon-Choles H, Vasylyeva TL, Pergola PE, Bhandari B, Bhandari K, Zhang JH, Wang W, Gorin Y, Barnes JL, Abboud HE. ZO-1 expression and phosphorylation in diabetic nephropathy. Diabetes 2006;55:894-900. PubMed
- 48. Antonetti DA, Barber AJ, Khin S, Lieth E, Tarbell JM, Gardner TW. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells: Penn State Retina Research Group. Diabetes 1998;47:1953-1959. ArticlePubMedPDF
- 49. Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes: the Penn State Retina Research Group. Invest Ophthalmol Vis Sci 2000;41:3561-3568. PubMed
- 50. Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 1999;99:649-659. ArticlePubMed
- 51. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 2000;11:4131-4142. ArticlePubMedPMC
- 52. Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 2002;156:1099-1111. PubMedPMC
- 53. Ben-Yosef T, Belyantseva IA, Saunders TL, Hughes ED, Kawamoto K, Van Itallie CM, Beyer LA, Halsey K, Gardner DJ, Wilcox ER, Rasmussen J, Anderson JM, Dolan DF, Forge A, Raphael Y, Camper SA, Friedman TB. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet 2003;12:2049-2061. ArticlePubMed
- 54. Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 2003;161:653-660. ArticlePubMedPMCPDF
- 55. Miyamoto T, Morita K, Takemoto D, Takeuchi K, Kitano Y, Miyakawa T, Nakayama K, Okamura Y, Sasaki H, Miyachi Y, Furuse M, Tsukita S. Tight junctions in Schwann cells of peripheral myelinated axons: a lesson from claudin-19-deficient mice. J Cell Biol 2005;169:527-538. PubMedPMC
- 56. Tamura A, Kitano Y, Hata M, Katsuno T, Moriwaki K, Sasaki H, Hayashi H, Suzuki Y, Noda T, Furuse M, Tsukita S, Tsukita S. Megaintestine in claudin-15-deficient mice. Gastroenterology 2008;134:523-534. ArticlePubMed
- 57. Nakano Y, Kim SH, Kim HM, Sanneman JD, Zhang Y, Smith RJ, Marcus DC, Wangemann P, Nessler RA, Banfi B. A claudin-9-based ion permeability barrier is essential for hearing. PLoS Genet 2009;5:e1000610ArticlePubMedPMC
- 58. Muto S, Hata M, Taniguchi J, Tsuruoka S, Moriwaki K, Saitou M, Furuse K, Sasaki H, Fujimura A, Imai M, Kusano E, Tsukita S, Furuse M. Claudin-2-deficient mice are defective in the leaky and cation-selective paracellular permeability properties of renal proximal tubules. Proc Natl Acad Sci U S A 2010;107:8011-8016. ArticlePubMedPMC
- 59. Tatum R, Zhang Y, Salleng K, Lu Z, Lin JJ, Lu Q, Jeansonne BG, Ding L, Chen YH. Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am J Physiol Renal Physiol 2010;298:F24-F34. ArticlePubMed
- 60. Will C, Breiderhoff T, Thumfart J, Stuiver M, Kopplin K, Sommer K, Gunzel D, Querfeld U, Meij IC, Shan Q, Bleich M, Willnow TE, Muller D. Targeted deletion of murine Cldn16 identifies extra- and intrarenal compensatory mechanisms of Ca2+ and Mg2+ wasting. Am J Physiol Renal Physiol 2010;298:F1152-F1161. ArticlePubMed
- 61. Hayashi D, Tamura A, Tanaka H, Yamazaki Y, Watanabe S, Suzuki K, Suzuki K, Sentani K, Yasui W, Rakugi H, Isaka Y, Tsukita S. Deficiency of claudin-18 causes paracellular H+ leakage, up-regulation of interleukin-1β, and atrophic gastritis in mice. Gastroenterology 2012;142:292-304. ArticlePubMed
- 62. Fujita H, Hamazaki Y, Noda Y, Oshima M, Minato N. Claudin-4 deficiency results in urothelial hyperplasia and lethal hydronephrosis. PLoS One 2012;7:e52272ArticlePubMedPMC
- 63. Wada M, Tamura A, Takahashi N, Tsukita S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013;144:369-380. ArticlePubMed
- 64. Gonzalez-Mariscal L, Hernandez S, Vega J. Inventions designed to enhance drug delivery across epithelial and endothelial cells through the paracellular pathway. Recent Pat Drug Deliv Formul 2008;2:145-176. ArticlePubMed
- 65. Deli MA. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim Biophys Acta 2009;1788:892-910. ArticlePubMed
- 66. Felinski EA, Antonetti DA. Glucocorticoid regulation of endothelial cell tight junction gene expression: novel treatments for diabetic retinopathy. Curr Eye Res 2005;30:949-957. ArticlePubMed
REFERENCES
Fig. 1Routes of transendothelial transport. A schematic representation of paracellular and transcellular routes for transport of blood-borne hormones and solutes to interstitial space of underlying tissue.
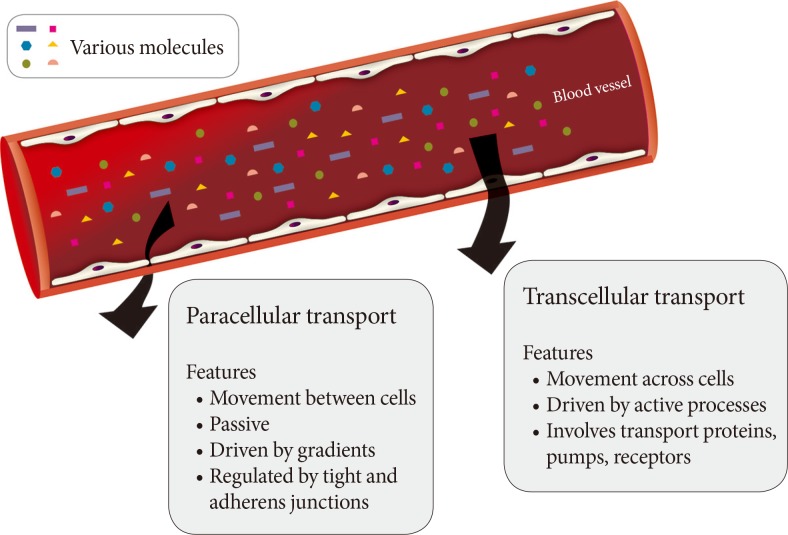
Fig. 2Diversity of paracellular transport characteristics in different vascular beds. The size and structure of tight junctions varies significantly between different tissues and in this figure we summarize this by defining three arbitrary categories of leaky, moderately tight and tight endothelia. The typical resistance values associated with these definitions and examples of tissues where each category of endothelium is characteristic is shown. N/A, not applicable.
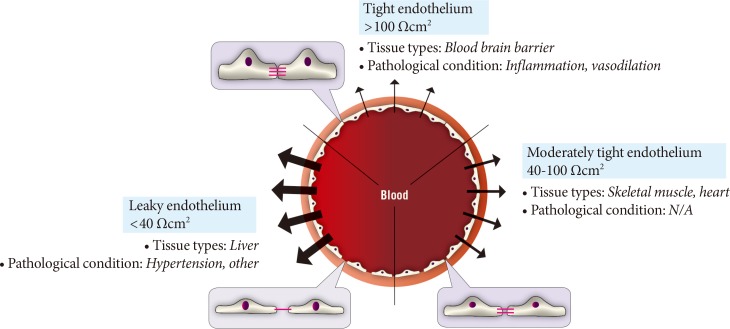
Fig. 3Paracellular movement of adiponectin. A conceptual model showing the potential significance of paracellular transendothelial movement of adiponectin. When vascular endothelium is leaky (left side) there is significant flux of all forms of adiponectin from bloodstream to interstitial space. However, as endothelium becomes tighter there is likely to be a gradient of decrease in high molecular weight or other multimeric forms of adiponectin whereas the smaller globular fragment of adiponectin may still be able to access underlying target tissue such as skeletal muscle. TJ, tight junctions; fAd, full length adiponectin; gAd, globular adiponectin.
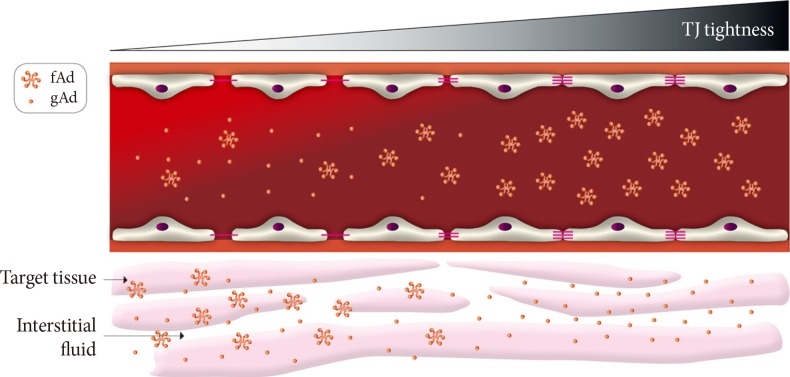
Figure & Data
References
Citations
Citations to this article as recorded by 
- Use of 2-dimensional cell monolayers and 3-dimensional microvascular networks on microfluidic devices shows that iron increases transendothelial adiponectin flux via inducing ROS production
Nanyoung Yoon, Seunggyu Kim, Hye Kyoung Sung, Thanh Q. Dang, Jessie S. Jeon, Gary Sweeney
Biochimica et Biophysica Acta (BBA) - General Subjects.2021; 1865(2): 129796. CrossRef - Adiponectin Synthesis, Secretion and Extravasation from Circulation to Interstitial Space
Simone C. da Silva Rosa, Meilian Liu, Gary Sweeney
Physiology.2021; 36(3): 134. CrossRef - Tracking adiponectin biodistribution via fluorescence molecular tomography indicates increased vascular permeability after streptozotocin-induced diabetes
Nanyoung Yoon, Keith Dadson, Thanh Dang, Teresa Chu, Nina Noskovicova, Boris Hinz, Adeline Raignault, Eric Thorin, Seunggyu Kim, Jessie S. Jeon, James Jonkman, Trevor D. McKee, Justin Grant, Jeffrey D. Peterson, Scott P. Kelly, Gary Sweeney
American Journal of Physiology-Endocrinology and Metabolism.2019; 317(5): E760. CrossRef - Overview of the Components of Cardiac Metabolism
Elizabeth A. Hausner, Susan A. Elmore, Xi Yang
Drug Metabolism and Disposition.2019; 47(6): 673. CrossRef - Emerging Roles of Vascular Endothelium in Metabolic Homeostasis
Xinchun Pi, Liang Xie, Cam Patterson
Circulation Research.2018; 123(4): 477. CrossRef - Transendothelial movement of adiponectin is restricted by glucocorticoids
Thanh Q Dang, Nanyoung Yoon, Helen Chasiotis, Emily C Dunford, Qilong Feng, Pingnian He, Michael C Riddell, Scott P Kelly, Gary Sweeney
Journal of Endocrinology.2017; 234(2): 101. CrossRef - Insulin access to skeletal muscle is impaired during the early stages of diet‐induced obesity
Josiane L. Broussard, Ana V.B. Castro, Malini Iyer, Rebecca L. Paszkiewicz, Isaac Asare Bediako, Lidia S. Szczepaniak, Edward W. Szczepaniak, Richard N. Bergman, Cathryn M. Kolka
Obesity.2016; 24(9): 1922. CrossRef - Temporal and Molecular Analyses of Cardiac Extracellular Matrix Remodeling following Pressure Overload in Adiponectin Deficient Mice
Keith Dadson, Subat Turdi, Stellar Boo, Boris Hinz, Gary Sweeney, Nikolaos Frangogiannis
PLOS ONE.2015; 10(4): e0121049. CrossRef
 PubReader
PubReader Cite
Cite