- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 36(5); 2012 > Article
-
ReviewPathophysiology Transcriptional Regulation of Pyruvate Dehydrogenase Kinase
- Ji Yun Jeong1, Nam Ho Jeoung2, Keun-Gyu Park1, In-Kyu Lee1
-
Diabetes & Metabolism Journal 2012;36(5):328-335.
DOI: https://doi.org/10.4093/dmj.2012.36.5.328
Published online: October 18, 2012
1Department of Internal Medicine, Kyungpook National University School of Medicine, Daegu, Korea.
2Department of Fundamental Medical & Pharmaceutical Sciences, Catholic University of Daegu, Daegu, Korea.
- Corresponding author: In-Kyu Lee. Division of Endocrinology and Metabolism, Department of Internal Medicine, Kyungpook National University School of Medicine, 130 Dongdeok-ro, Jung-gu, Daegu 700-721, Korea. leei@knu.ac.kr
Copyright © 2012 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The pyruvate dehydrogenase complex (PDC) activity is crucial to maintains blood glucose and ATP levels, which largely depends on the phosphorylation status by pyruvate dehydrogenase kinase (PDK) isoenzymes. Although it has been reported that PDC is phosphorylated and inactivated by PDK2 and PDK4 in metabolically active tissues including liver, skeletal muscle, heart, and kidney during starvation and diabetes, the precise mechanisms by which expression of PDK2 and PDK4 are transcriptionally regulated still remains unclear. Insulin represses the expression of PDK2 and PDK4 via phosphorylation of FOXO through PI3K/Akt signaling pathway. Several nuclear hormone receptors activated due to fasting or increased fat supply, including peroxisome proliferator-activated receptors, glucocorticoid receptors, estrogen-related receptors, and thyroid hormone receptors, also participate in the up-regulation of PDK2 and PDK4; however, the endogenous ligands that bind those nuclear receptors have not been identified. It has been recently suggested that growth hormone, adiponectin, epinephrine, and rosiglitazone also control the expression of PDK4 in tissue-specific manners. In this review, we discuss several factors involved in the expressional regulation of PDK2 and PDK4, and introduce current studies aimed at providing a better understanding of the molecular mechanisms that underlie the development of metabolic diseases such as diabetes.
- Selective fuel utilization depending on the fed-fast cycle is a crucial metabolic regulatory system in all mammals that is necessary to maintain a continuous and steady supply of ATP. In the fed state, increased availability of plasma glucose stimulates glucose oxidation and fatty acid synthesis. In the starved state, free fatty acids released from adipose tissue are selectively used for oxidative ATP generation in peripheral tissues and liver, and hepatic gluconeogenesis maintains the plasma glucose homeostasis. This regulation is mainly controlled by the activity of the pyruvate dehydrogenase complex (PDC), which regulates the entry of glycolytic products into the tricarboxylic acid cycle by catalyzing the oxidative decarboxylation of pyruvate to acetyl-CoA in mitochondria of mammalian cells. Although PDC is directly regulated by feedback inhibition by acetyl-CoA and NADH, both are end-products of oxidative decarboxylation of pyruvate and β-oxidation of free fatty acids, covalent modification of PDC via phosphorylation is recognized as critical for the long term regulation of PDC activity [1]. Pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP) are key regulators of PDC activity that act in a phosphorylation-dephosphorylation manner. Thus, the opposing activities of PDK and PDP regulate PDC activity.
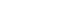
- Four PDK isoenzymes (PDK1, 2, 3, and 4) are known to be expressed in a tissue-specific manner in mammals. Of these, PDK2 and PDK4 have attracted the most interest due to the finding that their expressions are increased in many tissues during starvation and in diabetes. PDK2 is the most widely expressed isoform and particularly enriched in the liver and kidney [2,3]. The expression of PDK4 is dominantly increased in oxidative skeletal muscle, heart, lactating mammary gland, and liver [3-5]. As PDC activity is primarily controlled by acetyl-CoA and NADH, the activities of PDKs are also affected by the products and substrates of the catalyzing reaction. The mitochondrial levels of acetyl-CoA, NADH, and ATP increases PDK activity, while the levels of pyruvate, CoA-SH, NAD+, and ADP inhibit its activity (Fig. 1). This system controls the acute regulation of PDK activity during short-term food intake-deprivation cycles. The activity of PDK in metabolic disorders such as diabetes, heart disease, fatty liver, and long-term starvation is mainly controlled by transcriptional up-regulation of PDK.
INTRODUCTION
- PDK2 and PDK4 are highly expressed in liver, muscle, kidney, and heart tissue in starved and insulin-resistant animal models. Reduced insulin level or impaired insulin signaling contributes to the increase in both kinases. A remarkable increase in the amount of PDK4 mRNA occurs in the heart and skeletal muscle of streptozotocine-induced type 1 diabetic rat [4,6]. PDK4 is elevated in the skeletal muscle of insulin-resistant human subjects [7]. The negative regulatory effect of insulin on PDK4 expression was impaired in mice with acute insulin resistance induced by intra-lipid and lactate infusion, suggesting that insulin resistance may result in increase of PDK4 expression [8]. Likewise, insulin treatment reverse the increase of PDK4 and PDK2 expression in liver and skeletal muscle induced by dexamethasone [6,7].
- The role of insulin in the regulation of PDK expression is mediated in large part by the transcriptional activity of FOXO proteins. In particular, FOXO1 is activated by starvation and prevents hypoglycemia by inducing the transcription of gluconeogenic genes in the liver, such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), concurrent with PDK4 expression. To generate glucose, noncarbohydrate substrates such as pyruvate, lactate, and alanine must be conserved in the liver, which is dependent on the inactivation of PDC and a simultaneous increase in PDK activity. Fasting also activates FOXO1 in muscle, contributing to the induction of PDK and other genes involved in fatty acid utilization. Regulation of PDK by FOXO is reasonable to regulate fuel selection and maintain sufficient ATP levels through either glucose oxidation or fatty acid oxidation depending on the availability of energy source. Indeed, FOXO contributes to the regulation of PDK transcription by interacting with other nuclear hormone receptors such as the estrogen-related receptor (ERR), glucocorticoid receptor (GR), and peroxisome proliferator-activated receptor (PPAR). This is discussed in more detail below.
- The human PDK4 gene possesses three insulin response sequences (IRSs) that are binding sites for FOXO1 and FOXO3 [9]. Overexpression of constitutively active FOXO1 and FOXO3, which contain alanines at the three protein kinase B (Akt/PKB) phosphorylation sites, augmented both basal and dexamethasone-mediated increased expression of PDK4 in HepG2 cells; these augmentations were eliminated by mutation of the IRSs in the PDK4 promoter [9]. In mouse muscle and C2C12 cells, nutrient deprivation induced FOXO gene expression, which mediated the up-regulation of PDK4 gene expression [10]. Nahle et al. [11] reported that over-expression of CD36 in C2C12 cells, which facilitates fatty acid uptake in muscle, induced FOXO1 expression, thereby contributing to the up-regulation of muscle PDK4 expression.
- Although it is now well established that reduced insulin levels are responsible for the induction of PDK2, little is known about the PDK2 promoter. The insulin-mediated reduction of PDK2 mRNA expression in hepatoma cells might be associated with a decreased stability of the transcript [12]. We recently confirmed that PDK2 is dominantly expressed in the liver tissue of insulin-resistant animal models, including high fat dietinduced obese mice and db/db mice (unpublished data). Although the precise mechanism that mediates the up-regulation of PDK2 has not been elucidated, an increased fat supply in conjunction with insulin resistance may lead to an increased expression of PDK2 in the liver.
REGULATION OF PDK EXPRESSION BY INSULIN-FORKHEAD BOX O (FOXO) SIGNALING
- PPARs
- PPARs are a family of nuclear hormone receptors that function as transcription factors to regulate the expression of genes involved in metabolic pathways. Among the three main isoforms (PPARα, PPARβ/δ, and PPARγ), PPARα, an important adaptive regulator during prolonged fasting that promotes ketogenesis and fatty acid oxidation, is associated with increased expression of PDKs. Wu et al. [5] have reported a novel observation that WY-14,643, a PPAR-α agonist, dramatically induced both PDK4 mRNA and its protein in mouse skeletal muscle. Consequently, up-regulation of PDK4 by PPARα agonists, including fibrates and WY-14,643, has been investigated in several tissues such as heart, skeletal muscle, liver, and pancreatic β-cells [3,13-16]. Recent research shows that in skeletal muscle, pharmacologic activation of the liver X receptor (LXR) enhances PPARα-dependent up- and down-regulation of PDK4 in both fed and fasting conditions [17]. Although the clinical implication of LXR activation followed by a change in PDK4 expression, it has not been evaluated yet.
- The results of a study performed in human and mouse kidney cells using the PPARβ/δ agonist GW501516 suggest that PPARβ/δ lies upstream of PDK2, PDK3, and PDK4 [18]. In addition, it has been reported that PPARβ/δ induces FOXO1 expression via transcriptional regulation in muscle cell lines [11]. PPARγ is highly expressed in adipose tissue and regulates adipogenesis and glucose metabolism. Treatment with the PPARγ agonist rosiglitazone increased the amount of PDK4 in white adipose tissue, but not in liver or muscle tissue [19]. This will be discussed in detail in the section REGULATION OF PDK4 IN ADIPOSE TISSUE. In HEK293 cells, PPARγ ligands influence the mRNA expression of PDK3 and PDK4, but not PDK2 [18]. In addition, some evidence exists to suggest that PDK2 is not likely a direct target of PPARs in metabolically active tissues [3,18,19].
- It is of interest that free fatty acids and their derivatives have been implicated as endogenous PPAR ligands responsible for enhanced PDK expression in fasting and diabetes. Several reports demonstrate that long-chain fatty acids, such as palmitate and oleate, directly induce PDK4 mRNA in skeletal muscle and hepatoma cells [5,11,12]. Similarly, indirect supplies of fatty acids, such as increased levels of cardiac lipoprotein lipase and hepatic lipase, increase PDK4 mRNA expression by interacting with PPARα and PPARβ/δ [20,21]. When a hyperinsulinemic-euglycemic clamp was performed with healthy men, the suppression of PDK4 expression by insulin was attenuated by lipid infusion. In addition, this was independent of the activation of the Akt/PKB-mediated pathway, indicating that lipid accumulation is a more important factor than insulin signaling in the regulation of PDK4 expression in skeletal muscle [22]. On the contrary, insulin-mediated suppression of PDK4 expression in skeletal muscle was independent of its effect on plasma fatty acid levels, indicating that increased expression of PDK4 in starvation and diabetes may be due to insulin deficiency rather than to increased levels of fatty acids [23]. Further studies to identify the activators of PPARs responsible for regulation of PDK expression will provide insight into the role of PDK in metabolic disorders such as diabetes and insulin resistance.
- GR
- Dexamethasone treatment has been observed to markedly increase PDK4 expression in several tissues [12,24], suggesting that glucocorticoid is an important regulator of PDK4 expression in starvation and diabetes. A glucocorticoid response element (GRE) has been identified in the -824/-809 promoter region of the human PDK4 gene [9]. In addition, FOXO has been shown to participate in the induction of PDK4 by glucocorticoids via interaction with insulin response elements (IREs) located in a region near the GRE in the human PDK4 promoter [9]. Activation of protein kinase B-α, a component of intracellular insulin signaling, attenuated the dexamethasone-induced increase in PDK4 expression by interrupting the binding between FOXO and the IRE [9]. Insulin has also been shown to effectively decrease PDK4 expression augmented by dexamethasone in a similar manner [12]. Although dexamethasone treatment is reported to enlarge the lipoprotein lipase pool, as well as PDK4 expression, in the rat heart [24], whether the increased delivery of free fatty acids to the heart results in the augmentation of PDK4 expression is still controversial. Hyperglycemia, one of the clinical manifestations of Cushing syndrome, might be explained in part by the glucocorticoid-induced up-regulation of PDK4. It is not yet clear whether glucocorticoids regulate the expression of PDK2.
- ERRs
- ERRs are orphan nuclear receptors that are involved in the transcriptional regulation of cellular metabolic pathways. Of the three known isoforms, ERRα and ERRγ are expressed in metabolically active tissues such as skeletal muscle and liver. Several lines of evidence suggest that ERRα and ERRγ stimulate fatty acid oxidation through interaction with the PPARγ coactivator 1-α (PGC-1α) and gluconeogenesis via up-regulation of gluconeogenic genes such as PEPCK and G6P'ase, respectively. The roles played by ERRs in metabolic regulation suggest a relationship between ERRs and PDKs. ERRα has been shown to be involved in transcriptional activation of the PDK4 gene in C2C12 cells [25]. In that study, they showed that up-regulation of the PDK4 gene by ERRα was independent of PPARα, but was in partnership with PCG-1α. Up-regulation of PDK4 by ERRα was shown to be independent of PPARα, but dependent on a partnership with PCG-1α. Immediately following this study, the transcriptional regulation of PDK4 by ERRα and ERRγ in the liver was reported, which was associated with the recruitment of PCG-1α to the PDK4 promoter [26]. Both studies identified two ERR binding sites in the PDK4 promoter that are conserved in rat, mouse, and human PDK4 genes [25,26].
- Thyroid hormone receptors (TRs)
- The speculation that PDK is regulated by thyroid hormone (TH) arose from the observation that in hyperthyroidism, fatty acid oxidation is increased in various organs by altering the activity of CPT1, which is a major determinant of the rate of fatty acid oxidation [27,28]. In cultured hepatocytes and myocytes, triiodothyronine (T3) has been shown to enhance PDK activity [29,30]. Although the primary TH-responsive isoenzyme(s) in each tissue remain unknown, it is apparent that PDK2 and PDK4 are the predominant target genes of TH.
- Previous studies have shown that experimental hyperthyroidism results in increased PDK activity with enhanced expression of PDK4, but not PDK2, in heart tissue [28,31]. In addition, another study has reported that the expression of PDK2 is increased in rat heart tissue in hypothyroid conditions, whereas acute T3 treatment paradoxically elevated PDK2 levels [32]. In the former experiments, they used relatively long-term hyperthyroidism models, but in the latter, T3 was infused for only 60 minutes. These study results suggest the possibility that different mechanisms mediate TH-dependent regulation of PDK in heart tissue depending on the duration of treatment.
- In the liver, both PDK2 and PDK4 protein expression were enhanced in models of hyperthyroidism [3]. On the other hand, high fat diets, which increase the circulating lipid supply in a manner similar to hyperthyroidism, have been shown to enhance expression of PDK2 but not PDK4, in the liver, suggesting that up-regulation of PDK by TH in the liver might not be directly related to the supply of fatty acids. The transcriptional activity of TH is initiated by binding to the thyroid hormone receptor TR. The TR then binds to thyroid response elements (TRE) in conjunction with the recruitment of various coactivators. Two TREs in the rat PDK4 gene promoter have been identified, confirming that TH is able to regulate the transcription of PDK4 [33]. PGC-1α and CCAAT-enhancer-binding protein β (C/EBPβ) were discovered to be coactivators involved in the T3-mediated up-regulation of PDK4 [33,34]. Although the role of C/EBPβ has not been widely examined, it is well established that both TH and PGC-1α are linked to hepatic metabolism through the transcriptional regulation of gluconeogenic genes such as PEPCK. They also promote fatty acid oxidation, indicating that TH and PGC-1α have similar roles in the up-regulation of PDK [35,36]. Further study will be needed to determine the mechanism by which PDKs affect hepatic glucose-lipid metabolism in cooperation with transcriptional regulators, including TH, PGC-1α, and C/EBPβ.
REGULATION OF PDKS BY NUCLEAR HORMONE RECEPTORS
- Growth hormone (GH)
- As previously noted, fasting-induced hormones, such as glucocorticoids, have the potential to regulate PDK expression. GH is also a counter-regulatory hormone that is induced during fasting and participates in preventing hypoglycemia. The first study to describe the relationship between GH and PDK was published in 1974, and it showed that a GH polypeptide inhibited PDC activity in muscle [37]. However, subsequent data demonstrating that GH could increase PDC activity in rat hepatocytes, human mononuclear cells, and rat myocardium [38-40] were in contrast with recent studies that demonstrated that GH up-regulated PDK4 expression [41,42]. In one study, insulin could attenuate the ability of GH to induce PDK4 mRNA expression in adipose tissue, which is to be expected considering that GH plays roles as a counter-regulatory hormone in glucose metabolism. In addition, up-regulation of PDK4 by GH was mediated by STAT5A, which is a well-known downstream target of GH. It was recently established that GH also induces mRNA expression of PDK4 in the liver in a STAT5-dependent manner [42]. This induction was inhibited by treatment with metformin, which was accompanied by AMPK-mediated SHP activation. This novel observation of a GHSTAT5-PDK4 cascade in the liver may have clinical implications for the treatment of conditions with dysregulated hepatic metabolism associated with excess levels of GH, such as acromegaly.
- Krüppel-like factor 15 (KLF15)
- KLF15 is increased by glucocorticoid signaling during fasting and decreased by insulin signaling during feeding in hepatocytes. The enhanced expression during starvation is associated with its ability to increase the expression of gluconeogenic genes such as PEKCK. In this regard, we hypothesized that KLF15 would interfere with the transcription of PDK4 in liver tissue. As we had speculated, PDK4 promoter had the binding consensus motif for KFL15, and KLF15 could induce the mRNA expression of PDK4 in the liver cell line (unpublished data). This observation was confirmed by demonstrating that a mutation in the promoter region at the presumptive KFL15 binding site affected the expression of PDK4.
- Adiponectin and AMPK
- Adiponectin, which has been recognized as an insulin sensitizer in skeletal muscle, stimulates fat oxidation via the activation of AMPK. McAinch and Cameron-Smith [43] investigated the effect of adiponectin and AMPK activation on PDK4 gene expression in cultured skeletal muscle tissue from lean, obese, and obese diabetic humans. Both adiponectin and pharmacological activation of AMPK by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) decreased PDK4 expression in obese and diabetic samples, suggesting the possibility that the decrease in blood glucose mediated by adiponectin resulted from the down-regulation of PDK4 in skeletal muscle. Meanwhile, it was published that AMPK activation by hypoxia or AICAR with free fatty acid supply synergistically induced PDK4 [13].
OTHER FACTORS THAT REGULATE PDK EXPRESSION
- Compared to other metabolic regulatory organs, such as skeletal muscle, liver, and heart, a relatively small number of studies on the expression and regulatory mechanism of PDK in adipose tissue have been published. Triglyceride turnover in adipose tissue is a crucial metabolic process because it facilitates fatty acid utilization during fasting. Furthermore, the release of excessive amounts of plasma free fatty acids from adipose tissue is associated with insulin resistance [44]. These effects can be mediated via glyceroneogenesis, which occurs through the conversion of non-glucose molecules such as pyruvate to glycerol. This pathway begins with the conversion of pyruvate to oxaloacetate by pyruvate carboxylase (PC) in the mitochondria, suggesting that the ratio of PDC to PC activity is a key regulator of this pathway. It has been suggested that changes in PDC activity mediated by PDK4 expression could affect glyceroneogenesis in adipose tissue. Cadoudal and colleagues [19] showed that rosiglitazone increased PDK4 expression in a transcriptionally regulated manner in adipose tissue in an insulin-resistant animal model and in cultured adipocytes from humans and rats. In addition, they provided evidence that induced PDK4 played a role in white adipose tissue glyceroneogenesis, demonstrating that pyruvate is highly incorporated into neutral lipids by rosiglitazone and is reduced by both PDK inhibitors and siRNA of PDK4.
- Epinephrine has also been used to activate PDK4 in adipose tissue. Wan et al. [45] demonstrated that acute epinephrine treatment increased PDK4 mRNA in cultured adipose tissue and epididymal fat in rats, and that this increase was attenuated by blocking p38 mitogen-activated protein kinase activity. Similarly, a 2-hour swim exercise also leads to an increase in PDK4 mRNA levels in epididymal adipose tissue. They performed this experiment with obese, insulin-resistant rats [46]. Interestingly, the induction of PDK4 in adipose tissue by acute epinephrine injection was to a similar extent in lean and high fat diet (HFD) rats, whereas the activation of p38 was blunted in adipose tissue only in HFD rats. These findings demonstrated that increased PDK4 expression in adipose tissue induced by epinephrine or exercise is independent of p38. Although the transcription factor involved in PDK4 up-regulation by rosiglitazone or epinephrine remains unknown, these results indicate that PDK4 may play a pivotal role in regulating glucose-lipid metabolism in adipose tissue.
REGULATION OF PDK4 IN ADIPOSE TISSUE
- Exploring the transcriptional pathways involved in regulating PDK isoenzymes expression will lead to a better understanding of the molecular mechanisms that underlie the development of metabolic disease such as diabetes. This review summarizes recent progress in elucidating the transcriptional control of PDK, focusing on the PDK2 and PDK4 isoenzymes that play a role in metabolic pathways in insulin-responsive tissues (Fig. 2). Insulin and its downstream targets, such as FOXO, are major regulators of PDK expression. In addition, several nuclear receptors and hormones that also function in metabolic pathways coordinately participate in the transcriptional regulation of PDK.
- Studies that evaluate the efficacy of controlling PDC activity by down-regulating PDK expression with PDK inhibitors in animal models of metabolic disease, including diabetes, fatty liver, and obesity, will provide an opportunity to identify new therapeutic targets for the treatment of metabolic diseases.
CONCLUSIONS
-
Acknowledgements
- This study was supported by a grant from the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A111345).
ACKNOWLEDGMENTS
- 1. Sugden MC, Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem 2006;112:139-149. ArticlePubMed
- 2. Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA. Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch Biochem Biophys 2000;381:1-7. ArticlePubMed
- 3. Holness MJ, Bulmer K, Smith ND, Sugden MC. Investigation of potential mechanisms regulating protein expression of hepatic pyruvate dehydrogenase kinase isoforms 2 and 4 by fatty acids and thyroid hormone. Biochem J 2003;369(Pt 3):687-695. ArticlePubMedPMCPDF
- 4. Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem J 1998;329(Pt 1):197-201. ArticlePubMedPMCPDF
- 5. Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 1999;48:1593-1599. ArticlePubMedPDF
- 6. Stavinoha MA, Rayspellicy JW, Hart-Sailors ML, Mersmann HJ, Bray MS, Young ME. Diurnal variations in the responsiveness of cardiac and skeletal muscle to fatty acids. Am J Physiol Endocrinol Metab 2004;287:E878-E887. ArticlePubMed
- 7. Rosa G, Di Rocco P, Manco M, Greco AV, Castagneto M, Vidal H, Mingrone G. Reduced PDK4 expression associates with increased insulin sensitivity in postobese patients. Obes Res 2003;11:176-182. ArticlePubMed
- 8. Kim YI, Lee FN, Choi WS, Lee S, Youn JH. Insulin regulation of skeletal muscle PDK4 mRNA expression is impaired in acute insulin-resistant states. Diabetes 2006;55:2311-2317. ArticlePubMedPDF
- 9. Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes 2004;53:899-910. PubMed
- 10. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 2003;375(Pt 2):365-371. ArticlePubMedPMCPDF
- 11. Nahle Z, Hsieh M, Pietka T, Coburn CT, Grimaldi PA, Zhang MQ, Das D, Abumrad NA. CD36-dependent regulation of muscle FoxO1 and PDK4 in the PPAR delta/beta-mediated adaptation to metabolic stress. J Biol Chem 2008;283:14317-14326. PubMedPMC
- 12. Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes 2002;51:276-283. PubMed
- 13. Houten SM, Chegary M, Te Brinke H, Wijnen WJ, Glatz JF, Luiken JJ, Wijburg FA, Wanders RJ. Pyruvate dehydrogenase kinase 4 expression is synergistically induced by AMP-activated protein kinase and fatty acids. Cell Mol Life Sci 2009;66:1283-1294. ArticlePubMedPDF
- 14. Zungu M, Young ME, Stanley WC, Essop MF. Chronic treatment with the peroxisome proliferator-activated receptor alpha agonist Wy-14,643 attenuates myocardial respiratory capacity and contractile function. Mol Cell Biochem 2009;330:55-62. ArticlePubMedPDF
- 15. Wan J, Jiang L, Lu Q, Ke L, Li X, Tong N. Activation of PPARdelta up-regulates fatty acid oxidation and energy uncoupling genes of mitochondria and reduces palmitate-induced apoptosis in pancreatic beta-cells. Biochem Biophys Res Commun 2010;391:1567-1572. PubMed
- 16. Zhao Y, Okuyama M, Hashimoto H, Tagawa Y, Jomori T, Yang B. Bezafibrate induces myotoxicity in human rhabdomyosarcoma cells via peroxisome proliferator-activated receptor alpha signaling. Toxicol In Vitro 2010;24:154-159. PubMed
- 17. Caton PW, Holness MJ, Bishop-Bailey D, Sugden MC. PPARalpha-LXR as a novel metabolostatic signalling axis in skeletal muscle that acts to optimize substrate selection in response to nutrient status. Biochem J 2011;437:521-530. PubMed
- 18. Degenhardt T, Saramaki A, Malinen M, Rieck M, Vaisanen S, Huotari A, Herzig KH, Muller R, Carlberg C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J Mol Biol 2007;372:341-355. PubMed
- 19. Cadoudal T, Distel E, Durant S, Fouque F, Blouin JM, Collinet M, Bortoli S, Forest C, Benelli C. Pyruvate dehydrogenase kinase 4: regulation by thiazolidinediones and implication in glyceroneogenesis in adipose tissue. Diabetes 2008;57:2272-2279. PubMedPMC
- 20. Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, Kim JK, Zechner R, Goldberg IJ. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem 2004;279:25050-25057. ArticlePubMed
- 21. Brown JD, Oligino E, Rader DJ, Saghatelian A, Plutzky J. VLDL hydrolysis by hepatic lipase regulates PPARdelta transcriptional responses. PLoS One 2011;6:e21209ArticlePubMedPMC
- 22. Tsintzas K, Chokkalingam K, Jewell K, Norton L, Macdonald IA, Constantin-Teodosiu D. Elevated free fatty acids attenuate the insulin-induced suppression of PDK4 gene expression in human skeletal muscle: potential role of intramuscular long-chain acyl-coenzyme A. J Clin Endocrinol Metab 2007;92:3967-3972. ArticlePubMed
- 23. Lee FN, Zhang L, Zheng D, Choi WS, Youn JH. Insulin suppresses PDK-4 expression in skeletal muscle independently of plasma FFA. Am J Physiol Endocrinol Metab 2004;287:E69-E74. ArticlePubMed
- 24. Qi D, Pulinilkunnil T, An D, Ghosh S, Abrahani A, Pospisilik JA, Brownsey R, Wambolt R, Allard M, Rodrigues B. Single-dose dexamethasone induces whole-body insulin resistance and alters both cardiac fatty acid and carbohydrate metabolism. Diabetes 2004;53:1790-1797. ArticlePubMedPDF
- 25. Araki M, Motojima K. Identification of ERRalpha as a specific partner of PGC-1alpha for the activation of PDK4 gene expression in muscle. FEBS J 2006;273:1669-1680. ArticlePubMed
- 26. Zhang Y, Ma K, Sadana P, Chowdhury F, Gaillard S, Wang F, McDonnell DP, Unterman TG, Elam MB, Park EA. Estrogen-related receptors stimulate pyruvate dehydrogenase kinase isoform 4 gene expression. J Biol Chem 2006;281:39897-39906. ArticlePubMed
- 27. Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Stimulation of carnitine acylcarnitine translocase activity in heart mitochondria from hyperthyroid rats. FEBS Lett 1996;397:260-262. ArticlePubMedPDF
- 28. Priestman DA, Donald E, Holness MJ, Sugden MC. Different mechanisms underlie the long-term regulation of pyruvate dehydrogenase kinase (PDHK) by tri-iodothyronine in heart and liver. FEBS Lett 1997;419:55-57. ArticlePubMedPDF
- 29. Orfali KA, Fryer LG, Holness MJ, Sugden MC. Interactive effects of insulin and triiodothyronine on pyruvate dehydrogenase kinase activity in cardiac myocytes. J Mol Cell Cardiol 1995;27:901-908. ArticlePubMed
- 30. Sugden MC, Fryer LG, Priestman DA, Orfali KA, Holness MJ. Increased hepatic pyruvate dehydrogenase kinase activity in fed hyperthyroid rats: studies in vivo and with cultured hepatocytes. Mol Cell Endocrinol 1996;119:219-224. ArticlePubMed
- 31. Sugden MC, Langdown ML, Harris RA, Holness MJ. Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: role of thyroid hormone status and lipid supply. Biochem J 2000;352(Pt 3):731-738. ArticlePubMedPMCPDF
- 32. Hyyti OM, Ning XH, Buroker NE, Ge M, Portman MA. Thyroid hormone controls myocardial substrate metabolism through nuclear receptor-mediated and rapid posttranscriptional mechanisms. Am J Physiol Endocrinol Metab 2006;290:E372-E379. ArticlePubMed
- 33. Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by thyroid hormone: role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha). J Biol Chem 2010;285:2375-2385. PubMed
- 34. Attia RR, Sharma P, Janssen RC, Friedman JE, Deng X, Lee JS, Elam MB, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by CCAAT/enhancer-binding protein beta (C/EBPbeta). J Biol Chem 2011;286:23799-23807. PubMedPMC
- 35. Raboudi N, Arem R, Jones RH, Chap Z, Pena J, Chou J, Field JB. Fasting and postabsorptive hepatic glucose and insulin metabolism in hyperthyroidism. Am J Physiol 1989;256(1 Pt 1):E159-E166. ArticlePubMed
- 36. Zhang Y, Ma K, Song S, Elam MB, Cook GA, Park EA. Peroxisomal proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1 alpha) enhances the thyroid hormone induction of carnitine palmitoyltransferase I (CPT-I alpha). J Biol Chem 2004;279:53963-53971. PubMed
- 37. Aylward JH, Bornstein J, Gould MK, Hall S. Inhibition of muscle pyruvate dehydrogenase by a polypeptide from growth hormone. Biochem Biophys Res Commun 1974;59:57-61. ArticlePubMed
- 38. Clot JP, Benelli C, de Galle B, Postel-Vinay MC, Durand D, Desbuquois B. Effects of growth hormone on pyruvate dehydrogenase activity in intact rat liver and in isolated hepatocytes: comparison with insulin. Metabolism 1988;37:1101-1106. ArticlePubMed
- 39. Clot JP, Benelli C, Fouque F, Bernard R, Durand D, Postel-Vinay MC. Pyruvate dehydrogenase activity is stimulated by growth hormone (GH) in human mononuclear cells: a new tool to measure GH responsiveness in man. J Clin Endocrinol Metab 1992;74:1258-1262. ArticlePubMed
- 40. Seiva FR, Berbert CM, Souza GA, Rocha KK, Ebaid GM, Burneiko RC, Novelli EL. Energy expenditure, lipid profile, oxidative stress, and cardiac energy metabolism after growth hormone treatment in obese young rats. Horm Metab Res 2010;42:496-501. ArticlePubMed
- 41. White UA, Coulter AA, Miles TK, Stephens JM. The STAT5A-mediated induction of pyruvate dehydrogenase kinase 4 expression by prolactin or growth hormone in adipocytes. Diabetes 2007;56:1623-1629. ArticlePubMedPDF
- 42. Kim YD, Kim YH, Tadi S, Yu JH, Yim YH, Jeoung NH, Shong M, Hennighausen L, Harris RA, Lee IK, Lee CH, Choi HS. Metformin inhibits growth hormone-mediated hepatic pyruvate dehydrogenase kinase 4 gene expression through induction of orphan nuclear receptor small heterodimer partner. Diabetes 2012;61:2484-2494. PubMedPMC
- 43. McAinch AJ, Cameron-Smith D. Adiponectin decreases pyruvate dehydrogenase kinase 4 gene expression in obese- and diabetic-derived myotubes. Diabetes Obes Metab 2009;11:721-728. ArticlePubMed
- 44. Nye C, Kim J, Kalhan SC, Hanson RW. Reassessing triglyceride synthesis in adipose tissue. Trends Endocrinol Metab 2008;19:356-361. ArticlePubMed
- 45. Wan Z, Thrush AB, Legare M, Frier BC, Sutherland LN, Williams DB, Wright DC. Epinephrine-mediated regulation of PDK4 mRNA in rat adipose tissue. Am J Physiol Cell Physiol 2010;299:C1162-C1170. ArticlePubMed
- 46. Wan Z, Frier BC, Williams DB, Wright DC. Epinephrine induces PDK4 mRNA expression in adipose tissue from obese, insulin resistant rats. Obesity (Silver Spring) 2012;20:453-456. ArticlePubMedPDF
REFERENCES
Fig. 1Regulation of pyruvate dehydrogenase kinases (PDKs) by allosteric effectors. Among the products and substrates of pyruvate dehydrogenase complex (PDC), pyruvate, CoA, and nicotinamide adenine dinucleotide (NAD+) suppress PDK activity, while acetyl coenzyme A (acetyl-CoA) and NADH activate PDKs. ADP produced by the kinase reaction also inhibits PDK activity. TCA cycle, tricarboxylic acid cycle.
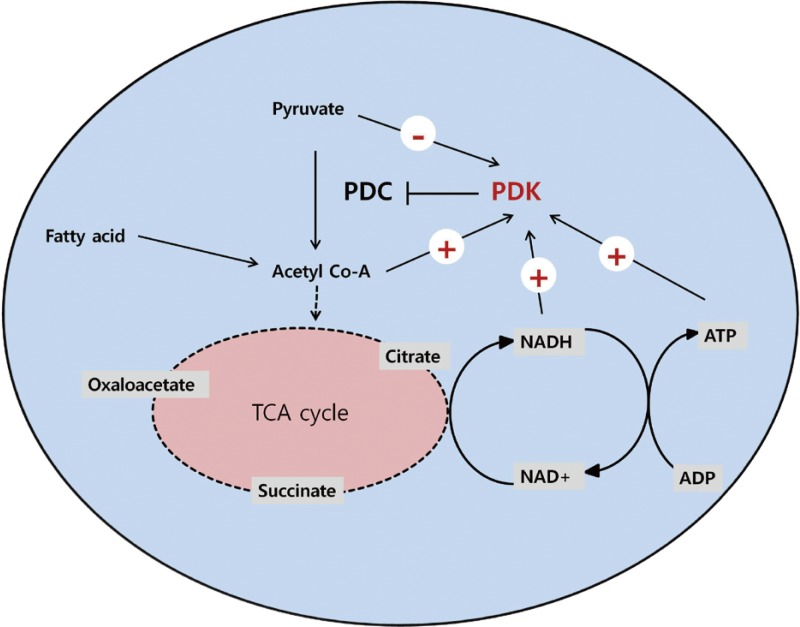
Fig. 2Schematic representation of transcriptional control of pyruvate dehydrogenase kinase 4 (PDK4) expression. Activation of Akt/PKB signaling by insulin phosphorylates forkhead box protein O (FOXO) and suppresses PDK4 expression. Several nuclear receptors, including estrogen-related receptor (ERR), peroxisome proliferator-activated receptor (PPAR), glucocorticoid receptor (GR), and thyroid hormone receptor (TR), participate in the transcriptional regulation of PDK4 and coordinate with coactivators, including peroxisome proliferator-activated receptor gamma coactivator 1-α (PCG-1α) and CCAAT/enhancer binding protein β (C/EBPβ).
Figure & Data
References
Citations
Citations to this article as recorded by 
- Molecules and targets of antidiabetic interest
Kavishankar Gawli, Kavya Sritha Bojja
Phytomedicine Plus.2024; 4(1): 100506. CrossRef - Transcriptome analysis reveals dysfunction of the endoplasmic reticulum protein processing in the sonic muscle of small yellow croaker (Larimichthys polyactis) following noise exposure
Xuguang Zhang, Xianming Tang, Jianan Xu, Yueping Zheng, Jun Lin, Huafeng Zou
Marine Environmental Research.2024; 194: 106299. CrossRef - Enhancing aortic valve drug delivery with PAR2-targeting magnetic nano-cargoes for calcification alleviation
Jinyong Chen, Tanchen Ren, Lan Xie, Haochang Hu, Xu Li, Miribani Maitusong, Xuhao Zhou, Wangxing Hu, Dilin Xu, Yi Qian, Si Cheng, Kaixiang Yu, Jian`an Wang, Xianbao Liu
Nature Communications.2024;[Epub] CrossRef - Natural Product-Based Glycolysis Inhibitors as a Therapeutic Strategy for Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer
Wonyoung Park, Jung Ho Han, Shibo Wei, Eun-Sun Yang, Se-Yun Cheon, Sung-Jin Bae, Dongryeol Ryu, Hwan-Suck Chung, Ki-Tae Ha
International Journal of Molecular Sciences.2024; 25(2): 807. CrossRef - 1,5-Anhydroglucitol promotes pre-B acute lymphocytic leukemia progression by driving glycolysis and reactive oxygen species formation
Huasu Zhu, Huixian Ma, Na Dong, Min Wu, Dong Li, Linghong Liu, Qing Shi, Xiuli Ju
BMC Cancer.2023;[Epub] CrossRef - The pyruvate dehydrogenase complex: Life’s essential, vulnerable and druggable energy homeostat
Peter W. Stacpoole, Charles E. McCall
Mitochondrion.2023; 70: 59. CrossRef - A Conceptual Approach for Examining Effects of the Adolescent Bone Marrow Milieu on MSC Phenotype
Sanjana Kannikeswaran, Daniel G Whitney, Maureen J Devlin, Ying Li, Michelle S Caird, Andrea I Alford
JBMR Plus.2023;[Epub] CrossRef - Targeting PDK2 rescues stress-induced impaired brain energy metabolism
Changshui Wang, Changmeng Cui, Pengfei Xu, Li Zhu, Hongjia Xue, Beibei Chen, Pei Jiang
Molecular Psychiatry.2023; 28(10): 4138. CrossRef - Inhibition of pyruvate dehydrogenase kinase 4 ameliorates kidney ischemia-reperfusion injury by reducing succinate accumulation during ischemia and preserving mitochondrial function during reperfusion
Chang Joo Oh, Min-Ji Kim, Ji-Min Lee, Dong Hun Kim, Il-Young Kim, Sanghee Park, Yeongmin Kim, Kyung-Bok Lee, Sang-Hee Lee, Chae Won Lim, Myeongjin Kim, Jung-Yi Lee, Haushabhau S. Pagire, Suvarna H. Pagire, Myung Ae Bae, Dipanjan Chanda, Themis Thoudam, Ah
Kidney International.2023; 104(4): 724. CrossRef - High glucose promotes benign prostatic hyperplasia by downregulating PDK4 expression
Pengyu Wei, Dongxu Lin, Changcheng Luo, Mengyang Zhang, Bolang Deng, Kai Cui, Zhong Chen
Scientific Reports.2023;[Epub] CrossRef - PDK4 facilitates fibroblast functions and diabetic wound healing through regulation of HIF‐1α protein stability and gene expression
Zhouji Ma, Ran Mo, Ping Yang, Youjun Ding, Hao Zhang, Zheng Dong, Yutong Chen, Qian Tan
The FASEB Journal.2023;[Epub] CrossRef - PDK4 rescues high-glucose-induced senescent fibroblasts and promotes diabetic wound healing through enhancing glycolysis and regulating YAP and JNK pathway
Zhouji Ma, Youjun Ding, Xiaofeng Ding, Haining Mou, Ran Mo, Qian Tan
Cell Death Discovery.2023;[Epub] CrossRef - CD36 deletion ameliorates diabetic kidney disease by restoring fatty acid oxidation and improving mitochondrial function
Huimin Niu, Xiayu Ren, Enxue Tan, Xing Wan, Yu Wang, Honghong Shi, Yanjuan Hou, Lihua Wang
Renal Failure.2023;[Epub] CrossRef - PDK4 Decrease Neuronal Apoptosis via Inhibiting ROS-ASK1/P38 Pathway in Early Brain Injury After Subarachnoid Hemorrhage
Xuan Gao, Yong-Yue Gao, Hui-Ying Yan, Guang-Jie Liu, Yan Zhou, Tao Tao, Ting-Ting Yue, Cong Pang, Xiang-Xin Chen, Sen Gao, Ling-Yun Wu, Chun-Hua Hang, Wei Li
Antioxidants & Redox Signaling.2022; 36(7-9): 505. CrossRef - microRNA-15b-5p shuttled by mesenchymal stem cell-derived extracellular vesicles protects podocytes from diabetic nephropathy via downregulation of VEGF/PDK4 axis
Tiantian Zhao, Qingsong Jin, Lili Kong, Dongdong Zhang, Yaqin Teng, Liangyan Lin, Xiaoyan Yao, Yongjun Jin, Minglong Li
Journal of Bioenergetics and Biomembranes.2022; 54(1): 17. CrossRef - Water-Extracted Prunella vulgaris Alleviates Endometriosis by Reducing Aerobic Glycolysis
Min Kyoung Cho, Ling Jin, Jung Ho Han, Jung-Suk Jin, Se-Yun Cheon, Su Shin, Sung-Jin Bae, Jang-Kyung Park, Ki-Tae Ha
Frontiers in Pharmacology.2022;[Epub] CrossRef - Coenzyme A-Dependent Tricarboxylic Acid Cycle Enzymes Are Decreased in Alzheimer’s Disease Consistent With Cerebral Pantothenate Deficiency
Crystal Sang, Sasha A. Philbert, Danielle Hartland, Richard. D Unwin, Andrew W. Dowsey, Jingshu Xu, Garth J. S. Cooper
Frontiers in Aging Neuroscience.2022;[Epub] CrossRef - Identification of candidate aberrant differentially methylated/expressed genes in asthma
Zongling Wang, Lizhi Wang, Lina Dai, Yanan Wang, Erhong Li, Shuyuan An, Fengliang Wang, Dan Liu, Wen Pan
Allergy, Asthma & Clinical Immunology.2022;[Epub] CrossRef - Innervation and electrical pulse stimulation — in vitro effects on human skeletal muscle cells
Tomaz Marš, Katarina Miš, Marija Meznarič, Sonja Prpar Mihevc, Vid Jan, Fred Haugen, Boris Rogelj, Arild C. Rustan, G. Hege Thoresen, Sergej Pirkmajer, Nataša Nikolić
Applied Physiology, Nutrition, and Metabolism.2021; 46(4): 299. CrossRef - Mitochondrial Dysfunction: Cause or Consequence of Vascular Calcification?
Kanchan Phadwal, Christina Vrahnas, Ian G. Ganley, Vicky E. MacRae
Frontiers in Cell and Developmental Biology.2021;[Epub] CrossRef - BCKDK regulates the TCA cycle through PDC in the absence of PDK family during embryonic development
Lia Heinemann-Yerushalmi, Lital Bentovim, Neta Felsenthal, Ron Carmel Vinestock, Nofar Michaeli, Sharon Krief, Alon Silberman, Marina Cohen, Shifra Ben-Dor, Ori Brenner, Rebecca Haffner-Krausz, Maxim Itkin, Sergey Malitsky, Ayelet Erez, Elazar Zelzer
Developmental Cell.2021; 56(8): 1182. CrossRef - Unique adaptations in neonatal hepatic transcriptome, nutrient signaling, and one-carbon metabolism in response to feeding ethyl cellulose rumen-protected methionine during late-gestation in Holstein cows
Valentino Palombo, Abdulrahman Alharthi, Fernanda Batistel, Claudia Parys, Jessie Guyader, Erminio Trevisi, Mariasilvia D’Andrea, Juan J. Loor
BMC Genomics.2021;[Epub] CrossRef - PDK2: An Underappreciated Regulator of Liver Metabolism
Benjamin L. Woolbright, Robert A. Harris
Livers.2021; 1(2): 82. CrossRef - Endocrine Influence on Cardiac Metabolism in Development and Regeneration
Niall Graham, Guo N Huang
Endocrinology.2021;[Epub] CrossRef - Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas
Kristina M. Cook, Han Shen, Kelly J. McKelvey, Harriet E. Gee, Eric Hau
International Journal of Molecular Sciences.2021; 22(14): 7265. CrossRef - Pyruvate dehydrogenase kinase 4‐mediated metabolic reprogramming is involved in rituximab resistance in diffuse large B‐cell lymphoma by affecting the expression of MS4A1/CD20
Duanfeng Jiang, Qiuyu Mo, Xiaoying Sun, Xiaotao Wang, Min Dong, Guozhen Zhang, Fangping Chen, Qiangqiang Zhao
Cancer Science.2021; 112(9): 3585. CrossRef - Skeletal muscle energy metabolism in obesity
Abel M. Mengeste, Arild C. Rustan, Jenny Lund
Obesity.2021; 29(10): 1582. CrossRef - Loss of prion protein control of glucose metabolism promotes neurodegeneration in model of prion diseases
Hélène Arnould, Vincent Baudouin, Anne Baudry, Luiz W. Ribeiro, Hector Ardila-Osorio, Mathéa Pietri, Cédric Caradeuc, Cynthia Soultawi, Declan Williams, Marjorie Alvarez, Carole Crozet, Fatima Djouadi, Mireille Laforge, Gildas Bertho, Odile Kellermann, Je
PLOS Pathogens.2021; 17(10): e1009991. CrossRef - Transcriptional and free radical responses to LVAD therapy
Kajari Dhar, Asmini KC, Fang Qiu, Hesham Basma, Krupa K. Savalia, Jocelyn Jones, Alexandra M. Moulton, Matthew C. Zimmerman, John Um, Daniel Anderson, Marshall Hyden, Brian D. Lowes
Translational Medicine Communications.2020;[Epub] CrossRef - Fine-tuning the metabolic rewiring and adaptation of translational machinery during an epithelial-mesenchymal transition in breast cancer cells
Tamara Fernández-Calero, Marcos Davyt, Karen Perelmuter, Cora Chalar, Giovana Bampi, Helena Persson, Juan Pablo Tosar, Völundur Hafstað, Hugo Naya, Carlos Rovira, Mariela Bollati-Fogolín, Ricardo Ehrlich, Gilles Flouriot, Zoya Ignatova, Mónica Marín
Cancer & Metabolism.2020;[Epub] CrossRef - Co-expression network analysis predicts a key role of microRNAs in the adaptation of the porcine skeletal muscle to nutrient supply
Emilio Mármol-Sánchez, Yuliaxis Ramayo-Caldas, Raquel Quintanilla, Tainã Figueiredo Cardoso, Rayner González-Prendes, Joan Tibau, Marcel Amills
Journal of Animal Science and Biotechnology.2020;[Epub] CrossRef - Stimulating pyruvate dehydrogenase complex reduces itaconate levels and enhances TCA cycle anabolic bioenergetics in acutely inflamed monocytes
Xuewei Zhu, David Long, Manal Zabalawi, Brian Ingram, Barbara K. Yoza, Peter W. Stacpoole, Charles E. McCall
Journal of Leukocyte Biology.2020; 107(3): 467. CrossRef - GPR30-Expressing Gastric Chief Cells Do Not Dedifferentiate But Are Eliminated via PDK-Dependent Cell Competition During Development of Metaplasia
Masahiro Hata, Hiroto Kinoshita, Yoku Hayakawa, Mitsuru Konishi, Mayo Tsuboi, Yukiko Oya, Ken Kurokawa, Yuki Hayata, Hayato Nakagawa, Keisuke Tateishi, Hiroaki Fujiwara, Yoshihiro Hirata, Daniel L. Worthley, Yuki Muranishi, Takahisa Furukawa, Shunsuke Kon
Gastroenterology.2020; 158(6): 1650. CrossRef - The pyruvate dehydrogenase kinase 2 (PDK2) is associated with conidiation, mycelial growth, and pathogenicity in Fusarium graminearum
Tao Gao, Dan He, Xin Liu, Fang Ji, Jianhong Xu, Jianrong Shi
Food Production, Processing and Nutrition.2020;[Epub] CrossRef - PDK2 Deficiency Prevents Ovariectomy-Induced Bone Loss in Mice by Regulating the RANKL-NFATc1 Pathway During Osteoclastogenesis
Ji-Min Lee, Min-Ji Kim, Sun Joo Lee, Byung-Gyu Kim, Je-Yong Choi, Seung Mi Lee, Hye Jin Ham, Jung-Min Koh, Jae-Han Jeon, In-Kyu Lee
Journal of Bone and Mineral Research.2020; 36(3): 553. CrossRef - Hemistepsin A suppresses colorectal cancer growth through inhibiting pyruvate dehydrogenase kinase activity
Ling Jin, Eun-Yeong Kim, Tae-Wook Chung, Chang Woo Han, So Young Park, Jung Ho Han, Sung-Jin Bae, Jong Rok Lee, Young Woo Kim, Se Bok Jang, Ki-Tae Ha
Scientific Reports.2020;[Epub] CrossRef - Neither Excessive Nitric Oxide Accumulation nor Acute Hyperglycemia Affects the N-Acetylaspartate Network in Wistar Rat Brain Cells
Marlena Zyśk, Piotr Pikul, Robert Kowalski, Krzysztof Lewandowski, Monika Sakowicz-Burkiewicz, Tadeusz Pawełczyk
International Journal of Molecular Sciences.2020; 21(22): 8541. CrossRef - PDK4 promotes vascular calcification by interfering with autophagic activity and metabolic reprogramming
Wen-Qi Ma, Xue-Jiao Sun, Yi Zhu, Nai-Feng Liu
Cell Death & Disease.2020;[Epub] CrossRef - Increased pyruvate dehydrogenase activity in skeletal muscle of growth-restricted ovine fetuses
Alexander L. Pendleton, Laurel R. Humphreys, Melissa A. Davis, Leticia E. Camacho, Miranda J. Anderson, Sean W. Limesand
American Journal of Physiology-Regulatory, Integrative and Comparative Physiology.2019; 317(4): R513. CrossRef - Geniposide Improves Glucose Homeostasis via Regulating FoxO1/PDK4 in Skeletal Muscle
Yan Li, Haiou Pan, Xuetong Zhang, Hui Wang, Shengnan Liu, Hui Zhang, Haifeng Qian, Li Wang, Hao Ying
Journal of Agricultural and Food Chemistry.2019; 67(16): 4483. CrossRef - The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury
A Ra Kho, Bo Young Choi, Song Hee Lee, Dae Ki Hong, Jeong Hyun Jeong, Beom Seok Kang, Dong Hyeon Kang, Kyoung-Ha Park, Jae Bong Park, Sang Won Suh
Cells.2019; 8(5): 405. CrossRef - Predictive factors for the development of diabetes in cancer patients treated with phosphatidylinositol 3-kinase inhibitors
Gyuri Kim, Myungeun Yoo, Min Hee Hong, Byung-Wan Lee, Eun Seok Kang, Bong-Soo Cha, Hye Ryun Kim, Yong-ho Lee, Byoung Chul Cho
Cancer Chemotherapy and Pharmacology.2019; 84(2): 405. CrossRef - Next generation sequencing of RNA reveals novel targets of resveratrol with possible implications for Canavan disease
Maja Dembic, Henriette S. Andersen, Jean Bastin, Thomas K. Doktor, Thomas J. Corydon, Jörn Oliver Sass, Alexandra Lopes Costa, Fatima Djouadi, Brage S. Andresen
Molecular Genetics and Metabolism.2019; 126(1): 64. CrossRef - PDK4 Augments ER–Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity
Themis Thoudam, Chae-Myeong Ha, Jaechan Leem, Dipanjan Chanda, Jong-Seok Park, Hyo-Jeong Kim, Jae-Han Jeon, Yeon-Kyung Choi, Suthat Liangpunsakul, Yang Hoon Huh, Tae-Hwan Kwon, Keun-Gyu Park, Robert A. Harris, Kyu-Sang Park, Hyun-Woo Rhee, In-Kyu Lee
Diabetes.2019; 68(3): 571. CrossRef - Membrane-initiated cortisol action modulates early pyruvate dehydrogenase kinase 2 (pdk2) expression in fish skeletal muscle
Jorge E. Aedo, Rodrigo Zuloaga, Sebastián Boltaña, Alfredo Molina, Juan Antonio Valdés
Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology.2019; 233: 24. CrossRef - Restoring mitochondrial biogenesis with metformin attenuates β-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis
Wen-Qi Ma, Xue-Jiao Sun, Ying Wang, Yi Zhu, Xi-Qiong Han, Nai-Feng Liu
Molecular and Cellular Endocrinology.2019; 479: 39. CrossRef - Changes in acetyl-CoA mediate Sik3-induced maturation of chondrocytes in endochondral bone formation
Azuma Kosai, Nanao Horike, Yoshiaki Takei, Akihiro Yamashita, Kaori Fujita, Takashi Kamatani, Noriyuki Tsumaki
Biochemical and Biophysical Research Communications.2019; 516(4): 1097. CrossRef - Enhancing cardiac glycolysis causes an increase in PDK4 content in response to short-term high-fat diet
Maria F. Newhardt, Albert Batushansky, Satoshi Matsuzaki, Zachary T. Young, Melinda West, Ngun Cer Chin, Luke I. Szweda, Michael Kinter, Kenneth M. Humphries
Journal of Biological Chemistry.2019; 294(45): 16831. CrossRef - Upregulated PDK4 expression is a sensitive marker of increased fatty acid oxidation
Ina Katrine Nitschke Pettersen, Deusdedit Tusubira, Hanan Ashrafi, Sissel Elisabeth Dyrstad, Lena Hansen, Xiao-Zheng Liu, Linn Iren Hodneland Nilsson, Nils Gunnar Løvsletten, Kjetil Berge, Hege Wergedahl, Bodil Bjørndal, Øystein Fluge, Ove Bruland, Arild
Mitochondrion.2019; 49: 97. CrossRef - Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis
Benjamin L. Woolbright, Ganeshkumar Rajendran, Robert A. Harris, John A. Taylor
Molecular Cancer Therapeutics.2019; 18(10): 1673. CrossRef - Clinical use of plasma lactate concentration. Part 1: Physiology, pathophysiology, and measurement
Patricia G. Rosenstein, Brett S. Tennent‐Brown, Dez Hughes
Journal of Veterinary Emergency and Critical Care.2018; 28(2): 85. CrossRef - SIRT6 deacetylase transcriptionally regulates glucose metabolism in heart
Danish Khan, Mohsen Sarikhani, Subhajit Dasgupta, Babukrishna Maniyadath, Anwit S. Pandit, Sneha Mishra, Faiz Ahamed, Abhinav Dubey, Nowrin Fathma, Hanudatta S. Atreya, Ullas Kolthur‐Seetharam, Nagalingam R. Sundaresan
Journal of Cellular Physiology.2018; 233(7): 5478. CrossRef - Metabolic Changes Associated With Muscle Expression of SOD1G93A
Gabriella Dobrowolny, Elisa Lepore, Martina Martini, Laura Barberi, Abigail Nunn, Bianca Maria Scicchitano, Antonio Musarò
Frontiers in Physiology.2018;[Epub] CrossRef - Effect of sex on glucose handling by adipocytes isolated from rat subcutaneous, mesenteric and perigonadal adipose tissue
Floriana Rotondo, Ana Cecilia Ho-Palma, Xavier Remesar, José Antonio Fernández-López, María del Mar Romero, Marià Alemany
PeerJ.2018; 6: e5440. CrossRef - Heat stress decreases metabolic flexibility in skeletal muscle of growing pigs
Lidan Zhao, Ryan P. McMillan, Guohao Xie, Samantha G. L. W. Giridhar, Lance H. Baumgard, Samer El-Kadi, Joshua Selsby, Jason Ross, Nicholas Gabler, Matthew W. Hulver, Robert P. Rhoads
American Journal of Physiology-Regulatory, Integrative and Comparative Physiology.2018; 315(6): R1096. CrossRef - Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism
Rong Ke, Qicao Xu, Cong Li, Lingyu Luo, Deqiang Huang
Cell Biology International.2018; 42(4): 384. CrossRef - Advanced glycation end products accelerate calcification in VSMCs through HIF-1α/PDK4 activation and suppress glucose metabolism
Yi Zhu, Wen-Qi Ma, Xi-Qiong Han, Ying Wang, Xin Wang, Nai-Feng Liu
Scientific Reports.2018;[Epub] CrossRef - Nε-carboxymethyl-lysine promotes calcium deposition in VSMCs via intracellular oxidative stress-induced PDK4 activation and alters glucose metabolism
Wen-Qi Ma, Xi-Qiong Han, Ying Wang, Xin Wang, Yi Zhu, Nai-Feng Liu
Oncotarget.2017; 8(68): 112841. CrossRef - Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury
Chang Joo Oh, Chae-Myeong Ha, Young-Keun Choi, Sungmi Park, Mi Sun Choe, Nam Ho Jeoung, Yang Hoon Huh, Hyo-Jeong Kim, Hee-Seok Kweon, Ji-min Lee, Sun Joo Lee, Jae-Han Jeon, Robert A. Harris, Keun-Gyu Park, In-Kyu Lee
Kidney International.2017; 91(4): 880. CrossRef - PPARβ/δ in human cancer
Rolf Müller
Biochimie.2017; 136: 90. CrossRef - The physiological and molecular response of Aurelia sp.1 under hypoxia
Guoshan Wang, Yu Zhen, Zhigang Yu, Yan Shi, Qing Zhao, Jianyan Wang, Tiezhu Mi
Scientific Reports.2017;[Epub] CrossRef - A Drosophila model of GDAP1 function reveals the involvement of insulin signalling in the mitochondria-dependent neuromuscular degeneration
Víctor López del Amo, Martina Palomino-Schätzlein, Marta Seco-Cervera, José Luis García-Giménez, Federico Vicente Pallardó, Antonio Pineda-Lucena, Máximo Ibo Galindo
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease.2017; 1863(3): 801. CrossRef - Western diet enhances intestinal tumorigenesis in Min/+ mice, associating with mucosal metabolic and inflammatory stress and loss of Apc heterozygosity
Mikael Niku, Anne-Maria Pajari, Laura Sarantaus, Essi Päivärinta, Markus Storvik, Anu Heiman-Lindh, Santeri Suokas, Minna Nyström, Marja Mutanen
The Journal of Nutritional Biochemistry.2017; 39: 126. CrossRef - Small heterodimer partner (SHP) deficiency protects myocardia from lipid accumulation in high fat diet-fed mice
Jung Hun Ohn, Ji Yeon Hwang, Min Kyong Moon, Hwa Young Ahn, Hwan Hee Kim, Young Do Koo, Kwang-Il Kim, Hyuk Jae Chang, Hye Seung Lee, Hak Chul Jang, Young Joo Park, Catherine Mounier
PLOS ONE.2017; 12(10): e0186021. CrossRef - Involvement of oxidative modification of proteins related to ATP synthesis in the left ventricles of hamsters with cardiomyopathy
Sahoko Ichihara, Yuka Suzuki, Jie Chang, Kentaro Kuzuya, Chisa Inoue, Yuki Kitamura, Shinji Oikawa
Scientific Reports.2017;[Epub] CrossRef - Early induction of pyruvate dehydrogenase kinase 4 by retinoic acids in adipocytes
Emilie Distel, Thomas Cadoudal, Martine Collinet, Edwards A. Park, Chantal Benelli, Sylvie Bortoli
Molecular Nutrition & Food Research.2017;[Epub] CrossRef - Oncogenic role of PDK4 in human colon cancer cells
D Leclerc, D N T Pham, N Lévesque, M Truongcao, W D Foulkes, C Sapienza, R Rozen
British Journal of Cancer.2017; 116(7): 930. CrossRef - Fasting and Glucagon Stimulate Gene Expression of Pyruvate Dehydrogenase Kinase 4 in Chickens
Kazuhisa Honda, Shoko Takagi, Kiyotaka Kurachi, Haruka Sugimoto, Takaoki Saneyasu, Hiroshi Kamisoyama
The Journal of Poultry Science.2017; 54(4): 292. CrossRef - Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer
Peter W Stacpoole
JNCI: Journal of the National Cancer Institute.2017;[Epub] CrossRef - Reference Gene Selection and Prednisolone Target Gene Expression in Adipose Tissues of Friesian Cattle
Sara Divari, Enrica Berio, Bartolomeo Biolatti, Francesca Tiziana Cannizzo
Journal of Agricultural and Food Chemistry.2017; 65(50): 11140. CrossRef - Histone Deacetylase Inhibitors Protect Against Pyruvate Dehydrogenase Dysfunction in Huntington's Disease
Luana Naia, Teresa Cunha-Oliveira, Joana Rodrigues, Tatiana R. Rosenstock, Ana Oliveira, Márcio Ribeiro, Catarina Carmo, Sofia I. Oliveira-Sousa, Ana I. Duarte, Michael R. Hayden, A. Cristina Rego
The Journal of Neuroscience.2017; 37(10): 2776. CrossRef - Black Adzuki Bean (Vigna angularis) Extract Protects Pancreatic β Cells and Improves Glucose Tolerance in C57BL/6J Mice Fed a High-Fat Diet
Mina Kim, Dae Keun Kim, Youn-Soo Cha
Journal of Medicinal Food.2016; 19(5): 442. CrossRef - Inflammation increases pyruvate dehydrogenase kinase 4 (PDK4) expression via the Jun N-Terminal Kinase (JNK) pathway in C2C12 cells
Hana Park, Nam Ho Jeoung
Biochemical and Biophysical Research Communications.2016; 469(4): 1049. CrossRef - Mechanisms of Vascular Calcification: The Pivotal Role of Pyruvate Dehydrogenase Kinase 4
Jaechan Leem, In-Kyu Lee
Endocrinology and Metabolism.2016; 31(1): 52. CrossRef - Molecular Mechanisms of Obesity-Induced Osteoporosis and Muscle Atrophy
Bipradas Roy, Mary E. Curtis, Letimicia S. Fears, Samuel N. Nahashon, Hugh M. Fentress
Frontiers in Physiology.2016;[Epub] CrossRef - MiR-155 Enhances Insulin Sensitivity by Coordinated Regulation of Multiple Genes in Mice
Xiaolin Lin, Yujuan Qin, Junshuang Jia, Taoyan Lin, Xia Lin, Li Chen, Hui Zeng, Yanjiang Han, Lihong Wu, Shun Huang, Meng Wang, Shenhao Huang, Raoying Xie, Liqi Liang, Yu Liu, Ruiyu Liu, Tingting Zhang, Jing Li, Shengchun Wang, Penghui Sun, Wenhua Huang,
PLOS Genetics.2016; 12(10): e1006308. CrossRef - Transforming Growth Factor β Mediates Drug Resistance by Regulating the Expression of Pyruvate Dehydrogenase Kinase 4 in Colorectal Cancer
Yang Zhang, Yi Zhang, Liying Geng, Haowei Yi, Wei Huo, Geoffrey Talmon, Yeong C. Kim, San Ming Wang, Jing Wang
Journal of Biological Chemistry.2016; 291(33): 17405. CrossRef - Metabolic Response to Heat Stress in Late-Pregnant and Early Lactation Dairy Cows: Implications to Liver-Muscle Crosstalk
Franziska Koch, Ole Lamp, Mehdi Eslamizad, Joachim Weitzel, Björn Kuhla, Andrew C. Gill
PLOS ONE.2016; 11(8): e0160912. CrossRef - Short chain acyl-CoA dehydrogenase deficiency and short-term high-fat diet perturb mitochondrial energy metabolism and transcriptional control of lipid-handling in liver
Sujoy Ghosh, Claudia Kruger, Shawna Wicks, Jacob Simon, K. Ganesh Kumar, William D. Johnson, Randall L. Mynatt, Robert C. Noland, Brenda K. Richards
Nutrition & Metabolism.2016;[Epub] CrossRef - Metabolic reprogramming by the pyruvate dehydrogenase kinase–lactic acid axis: Linking metabolism and diverse neuropathophysiologies
Mithilesh Kumar Jha, In-Kyu Lee, Kyoungho Suk
Neuroscience & Biobehavioral Reviews.2016; 68: 1. CrossRef - 1α,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells
Zachary C. Ryan, Theodore A. Craig, Clifford D. Folmes, Xuewei Wang, Ian R. Lanza, Niccole S. Schaible, Jeffrey L. Salisbury, K. Sreekumaran Nair, Andre Terzic, Gary C. Sieck, Rajiv Kumar
Journal of Biological Chemistry.2016; 291(3): 1514. CrossRef - Janus and PI3-kinases mediate glucocorticoid resistance in activated chronic leukemia cells
Sina Oppermann, Avery J. Lam, Stephanie Tung, Yonghong Shi, Lindsay McCaw, Guizhei Wang, Jarkko Ylanko, Brian Leber, David Andrews, David E. Spaner
Oncotarget.2016; 7(45): 72608. CrossRef - Energy metabolic disorder is a major risk factor in severe influenza virus infection: Proposals for new therapeutic options based on animal model experiments
Hiroshi Kido, Irene L. Indalao, Hyejin Kim, Takashi Kimoto, Satoko Sakai, Etsuhisa Takahashi
Respiratory Investigation.2016; 54(5): 312. CrossRef - Fusarium graminearum pyruvate dehydrogenase kinase 1 (FgPDK1) Is Critical for Conidiation, Mycelium Growth, and Pathogenicity
Tao Gao, Jian Chen, Zhiqi Shi, Yin-Won Lee
PLOS ONE.2016; 11(6): e0158077. CrossRef - Cardiac Ryanodine Receptor (Ryr2)-mediated Calcium Signals Specifically Promote Glucose Oxidation via Pyruvate Dehydrogenase
Michael J. Bround, Rich Wambolt, Haoning Cen, Parisa Asghari, Razvan F. Albu, Jun Han, Donald McAfee, Marc Pourrier, Nichollas E. Scott, Lubos Bohunek, Jerzy E. Kulpa, S. R. Wayne Chen, David Fedida, Roger W. Brownsey, Christoph H. Borchers, Leonard J. Fo
Journal of Biological Chemistry.2016; 291(45): 23490. CrossRef - Pyruvate Dehydrogenase Kinases: Therapeutic Targets for Diabetes and Cancers
Nam Ho Jeoung
Diabetes & Metabolism Journal.2015; 39(3): 188. CrossRef - Novel molecular mechanisms involved in hormonal regulation of lactate production in Sertoli cells
Mariana Regueira, Silvana Lucía Artagaveytia, María Noel Galardo, Eliana Herminia Pellizzari, Selva Beatriz Cigorraga, Silvina Beatriz Meroni, María Fernanda Riera
REPRODUCTION.2015; 150(4): 311. CrossRef - Influence of dietary fatty acids on differentiation of human stromal vascular fraction preadipocytes
Anna Polus, Beata Kiec-Wilk, Urszula Razny, Anna Gielicz, Gerd Schmitz, Aldona Dembinska-Kiec
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids.2015; 1851(9): 1146. CrossRef - A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis
Lavinia Palamiuc, Anna Schlagowski, Shyuan T Ngo, Aurelia Vernay, Sylvie Dirrig‐Grosch, Alexandre Henriques, Anne‐Laurence Boutillier, Joffrey Zoll, Andoni Echaniz‐Laguna, Jean‐Philippe Loeffler, Frédérique René
EMBO Molecular Medicine.2015; 7(5): 526. CrossRef - A genetic perspective on rapid evolution in cane toads (Rhinella marina)
Lee A. Rollins, Mark F. Richardson, Richard Shine
Molecular Ecology.2015; 24(9): 2264. CrossRef - Transcriptome profiling of brown adipose tissue during cold exposure reveals extensive regulation of glucose metabolism
Qin Hao, Rachita Yadav, Astrid L. Basse, Sidsel Petersen, Si B. Sonne, Simon Rasmussen, Qianhua Zhu, Zhike Lu, Jun Wang, Karine Audouze, Ramneek Gupta, Lise Madsen, Karsten Kristiansen, Jacob B. Hansen
American Journal of Physiology-Endocrinology and Metabolism.2015; 308(5): E380. CrossRef - Pyruvate Dehydrogenase Kinase 4 Promotes Vascular Calcification via SMAD1/5/8 Phosphorylation
Sun Joo Lee, Ji Yun Jeong, Chang Joo Oh, Sungmi Park, Joon-Young Kim, Han-Jong Kim, Nam Doo Kim, Young-Keun Choi, Ji-Yeon Do, Younghoon Go, Chae-Myeong Ha, Je-Yong Choi, Seung Huh, Nam Ho Jeoung, Ki-Up Lee, Hueng-Sik Choi, Yu Wang, Keun-Gyu Park, Robert A
Scientific Reports.2015;[Epub] CrossRef - Limited capacity for glucose oxidation in fetal sheep with intrauterine growth restriction
Laura D. Brown, Paul J. Rozance, Jennifer L. Bruce, Jacob E. Friedman, William W. Hay, Stephanie R. Wesolowski
American Journal of Physiology-Regulatory, Integrative and Comparative Physiology.2015; 309(8): R920. CrossRef - The long‐acting β2‐adrenoceptor agonist, indacaterol, enhances glucocorticoid receptor‐mediated transcription in human airway epithelial cells in a gene‐ and agonist‐dependent manner
T Joshi, M Johnson, R Newton, M A Giembycz
British Journal of Pharmacology.2015; 172(10): 2634. CrossRef - The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism
Jianrong Lu, Ming Tan, Qingsong Cai
Cancer Letters.2015; 356(2): 156. CrossRef - The Role of Pyruvate Dehydrogenase Kinase in Diabetes and Obesity
In-Kyu Lee
Diabetes & Metabolism Journal.2014; 38(3): 181. CrossRef - Rapid in Vitro Metabolism of the Flame Retardant Triphenyl Phosphate and Effects on Cytotoxicity and mRNA Expression in Chicken Embryonic Hepatocytes
Guanyong Su, Doug Crump, Robert J. Letcher, Sean W. Kennedy
Environmental Science & Technology.2014; 48(22): 13511. CrossRef - Low force contractions induce fatigue consistent with muscle mRNA expression in people with spinal cord injury
Michael A. Petrie, Manish Suneja, Elizabeth Faidley, Richard K. Shields
Physiological Reports.2014; 2(2): e00248. CrossRef - Diisopropylamine Dichloroacetate, a Novel Pyruvate Dehydrogenase Kinase 4 Inhibitor, as a Potential Therapeutic Agent for Metabolic Disorders and Multiorgan Failure in Severe Influenza
Kazuhiko Yamane, Irene L. Indalao, Junji Chida, Yoshikazu Yamamoto, Masaaki Hanawa, Hiroshi Kido, Amy Lynn Adamson
PLoS ONE.2014; 9(5): e98032. CrossRef - Fatty Acid Transport Protein 1 (FATP1) Localizes in Mitochondria in Mouse Skeletal Muscle and Regulates Lipid and Ketone Body Disposal
Maria Guitart, Óscar Osorio-Conles, Thais Pentinat, Judith Cebrià, Judit García-Villoria, David Sala, David Sebastián, Antonio Zorzano, Antonia Ribes, Josep C. Jiménez-Chillarón, Celia García-Martínez, Anna M. Gómez-Foix, Cedric Moro
PLoS ONE.2014; 9(5): e98109. CrossRef - CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism
Blake C. Ellis, Lloyd D. Graham, Peter L. Molloy
Biochimica et Biophysica Acta (BBA) - Molecular Cell Research.2014; 1843(2): 372. CrossRef - DLK1/PREF1 regulates nutrient metabolism and protects from steatosis
Marika Charalambous, Simao Teixeira Da Rocha, Elizabeth Jane Radford, Gema Medina-Gomez, Scott Curran, Scarlett B. Pinnock, Sacri R. Ferrón, Antonio Vidal-Puig, Anne C. Ferguson-Smith
Proceedings of the National Academy of Sciences.2014; 111(45): 16088. CrossRef - The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility
Shuai Zhang, Matthew W Hulver, Ryan P McMillan, Mark A Cline, Elizabeth R Gilbert
Nutrition & Metabolism.2014;[Epub] CrossRef - Time‐dependent effects of the flame retardant tris(1,3‐dichloro‐2‐propyl) phosphate (TDCPP) on mRNA expression, in vitro and in ovo, reveal optimal sampling times for rapidly metabolized compounds
Amani Farhat, Doug Crump, Emily Porter, Suzanne Chiu, Robert J. Letcher, Guanyong Su, Sean W. Kennedy
Environmental Toxicology and Chemistry.2014; 33(12): 2842. CrossRef - Chickens from lines selected for high and low body weight show differences in fatty acid oxidation efficiency and metabolic flexibility in skeletal muscle and white adipose tissue
S Zhang, R P McMillan, M W Hulver, P B Siegel, L H Sumners, W Zhang, M A Cline, E R Gilbert
International Journal of Obesity.2014; 38(10): 1374. CrossRef - Pathway-selective Insulin Resistance and Metabolic Disease: The Importance of Nutrient Flux
Yolanda F. Otero, John M. Stafford, Owen P. McGuinness
Journal of Biological Chemistry.2014; 289(30): 20462. CrossRef - Remodeling of Oxidative Energy Metabolism by Galactose Improves Glucose Handling and Metabolic Switching in Human Skeletal Muscle Cells
Eili Tranheim Kase, Nataša Nikolić, Siril Skaret Bakke, Kaja Kamilla Bogen, Vigdis Aas, G. Hege Thoresen, Arild Christian Rustan, Darcy Johannsen
PLoS ONE.2013; 8(4): e59972. CrossRef - Association of Estrogen Receptor α GenesPvuII andXbaI Polymorphisms with Type 2 Diabetes Mellitus in the Inpatient Population of a Hospital in Southern Iran
Farzaneh Mohammadi, Mohammad Pourahmadi, Mohadeseh Mosalanejad, Houshang Jamali, Mohamed Amin Ghobadifar, Saeideh Erfanian
Diabetes & Metabolism Journal.2013; 37(4): 270. CrossRef - The orphan nuclear receptors in cancer and diabetes
Harmit S. Ranhotra
Journal of Receptors and Signal Transduction.2013; 33(4): 207. CrossRef - Genes with Aberrant Expression in Murine Preneoplastic Intestine Show Epigenetic and Expression Changes in Normal Mucosa of Colon Cancer Patients
Daniel Leclerc, Nancy Lévesque, Yuanhang Cao, Liyuan Deng, Qing Wu, Jasmine Powell, Carmen Sapienza, Rima Rozen
Cancer Prevention Research.2013; 6(11): 1171. CrossRef
 PubReader
PubReader Cite
Cite