Mitochondrial Stress and Mitokines: Therapeutic Perspectives for the Treatment of Metabolic Diseases
Article information

Abstract
Mitochondrial stress and the dysregulated mitochondrial unfolded protein response (UPRmt) are linked to various diseases, including metabolic disorders, neurodegenerative diseases, and cancer. Mitokines, signaling molecules released by mitochondrial stress response and UPRmt, are crucial mediators of inter-organ communication and influence systemic metabolic and physiological processes. In this review, we provide a comprehensive overview of mitokines, including their regulation by exercise and lifestyle interventions and their implications for various diseases. The endocrine actions of mitokines related to mitochondrial stress and adaptations are highlighted, specifically the broad functions of fibroblast growth factor 21 and growth differentiation factor 15, as well as their specific actions in regulating inter-tissue communication and metabolic homeostasis. Finally, we discuss the potential of physiological and genetic interventions to reduce the hazards associated with dysregulated mitokine signaling and preserve an equilibrium in mitochondrial stress-induced responses. This review provides valuable insights into the mechanisms underlying mitochondrial regulation of health and disease by exploring mitokine interactions and their regulation, which will facilitate the development of targeted therapies and personalized interventions to improve health outcomes and quality of life.
INTRODUCTION
Mitochondria are essential for cellular energy metabolism. They are often referred to as the “powerhouses” of the cell because they are responsible for generating most of the adenosine triphosphate (ATP) molecules that cells use as a source of energy [1,2]. In addition to ATP production, mitochondria also play a critical role in other aspects of cellular metabolism, including fatty acid metabolism, amino acid metabolism, and the synthesis of heme and iron-sulfur clusters [3]. They are also involved in the regulation of cellular signaling pathways, cell death, and production of reactive oxygen species (ROS) [4-6], which can have both beneficial and detrimental effects on cells depending on the context [7,8]. Given their central role in cellular energy metabolism and other metabolic processes, dysfunction of mitochondria has been implicated in a wide range of diseases, including metabolic disorders, neurodegenerative diseases, and cancer [9-11].
Mitochondria are not simply static organelles within cells but form dynamic networks that exchange molecules and information both among themselves and with other organelles and the cytoplasm. This communication between mitochondria is increasingly recognized as being able to extend beyond cellular boundaries and even across different tissues [12-14]. However, the precise roles of mitochondria in inter-tissue communication and the implications for health and disease are not fully understood. One fascinating example of inter-tissue crosstalk partially mediated by mitochondria is the communication of exercise signals from skeletal muscles to various organs [15-17], including the brain [18,19]. This area of research is an exciting and rapidly developing field, with the potential to provide numerous insights into the mechanisms by which mitochondria influence systemic metabolic and physiological processes.
The mitochondrial stress response (MSR) and mitochondrial unfolded protein response (UPRmt) have also been investigated as potential cellular mechanisms underlying inter-organ communication and metabolic homeostasis [20,21]. The MSR is a process by which cells respond to stressors that disrupt mitochondrial function, such as oxidative stress and mitochondrial DNA damage [22,23]. The response involves the activation of signaling pathways that trigger changes in gene expression and protein synthesis to restore mitochondrial function and maintain cellular homeostasis [11,24]. Recent studies have suggested that the MSR may also play a role in inter-tissue communication, with mitochondria releasing signaling molecules and cell non-autonomous factors including mitokines, which can influence metabolic and physiological processes in other organs and tissues [2]. The UPRmt is another important cellular mechanism for maintaining mitochondrial homeostasis [25]. It involves the activation of signaling pathways that respond to the accumulation of unfolded or misfolded proteins within mitochondria, which can disrupt mitochondrial function and lead to cellular stress [26]. The UPRmt promotes the degradation of damaged proteins and the synthesis of new proteins to restore mitochondrial function and maintain cellular homeostasis [27,28]. Recent studies have also suggested that the UPRmt may be involved in inter-tissue communication, with mitochondria releasing signaling molecules that can influence metabolic processes in other organs and tissues [2]. Overall, these cellular mechanisms that maintain mitochondrial homeostasis, the MSR and UPRmt, are increasingly recognized as having important roles in inter-organ communication and metabolic homeostasis.
In recent years, research of mitokines and their actions has advanced significantly, shedding light on the important roles that these signaling molecules play in mediating inter-organ communication and influencing systemic metabolic and physiological processes [29]. The present review aims to provide a comprehensive overview of the current state of knowledge about mitokines, including their regulation by exercise and other lifestyle interventions, and their implications for aging. However, despite the progress that has been made in this area, many important questions remain unanswered. In particular, there is still much to be learned about how mitokines interact with other mitochondrial communication channels and the mechanisms by which they mediate inter-tissue crosstalk. Furthermore, while the beneficial effects of mitokine signaling on aging and disease prevention are becoming increasingly apparent, the risks associated with dysregulated mitokine signaling and its potential to facilitate metabolic diseases must also be carefully considered.
In this review, our objective is to provide a focused assessment of the existing literature pertaining to mitokine-mediated communication, with a specific emphasis on the interactions among MSR pathways, the UPRmt, and additional signaling mechanisms. Furthermore, we will examine the feasibility of employing physiological and genetic interventions to preserve an equilibrium in mitochondrial stress-induced communication and reduce the hazards linked to aberrant mitokine signaling. In summary, this review aims to provide valuable insights into the mechanisms underlying inter-tissue communication and the potential application of mitokine signaling as a therapeutic target for metabolic disorders.
INTER-TISSUE AND MULTI-ORGAN ORCHESTRATION IN RESPONSE TO MITOCHONDRIAL STRESS
Mitochondrial insults, such as oxidative stress, unfolded proteins, and impairment of the electron transport system, can all lead to perturbation of mitochondrial protein import, which triggers a MSR in mammalian cells. The MSR coordinates an array of adaptive responses, including activation of the UPRmt, to restore mitochondrial function and maintain cellular homeostasis. The UPRmt is a conserved adaptive response mechanism that promotes mitochondrial protein quality control by activating specific signaling pathways in response to mitochondrial stress [30]. When unfolded or misfolded proteins accumulate within the mitochondrial matrix [31,32], the UPRmt is activated to promote the degradation of damaged proteins and the synthesis of new proteins in order to restore mitochondrial function and maintain cellular homeostasis [27]. The intricate cellular mechanisms underlying the MSR and UPRmt are critical determinants of the aging process and are increasingly being recognized as essential for maintaining cellular homeostasis [33-35]. The UPRmt is a retrograde response that signals proteotoxic stress and triggers protective adaptations, such as metabolic reprogramming and epigenetic remodeling [36,37]. This response is initiated by the accumulation of unfolded or misfolded proteins within the mitochondrial matrix, which triggers a signaling cascade that ultimately leads to changes in gene expression and protein synthesis in order to restore mitochondrial function and maintain cellular homeostasis. The MSR and UPRmt pathways have multiple targets, including the regulation of mitochondrial protein import, mitochondrial and cytoplasmic proteostasis, recovery of oxidative phosphorylation, and clearance of defective mitochondria through mitophagy [20,38-41]. Mitophagy, a selective autophagic process involved in the degradation and removal of damaged or dysfunctional mitochondria, plays a critical role in regulating mitochondrial morphology in response to metabolic states. This dynamic regulation of mitochondrial morphology through mitophagy, fusion, and fission allows mitochondria to adapt their structure and function to varying metabolic demands, ensuring cellular health and efficient energy production. Additionally, other cellular mechanisms, such as eukaryotic initiation factor 2α (eIF2α; translation initiation factor)-mediated reduction of global protein synthesis and the heat shock response, also contribute to cellular stress adaptations [42,43].
Both the MSR and UPRmt have been proposed to be forms of hormesis or mitohormesis. Hormesis refers to a beneficial adaptive response to low levels of stress or damage that can activate cellular repair mechanisms and promote cellular stress resistance. Mitohormesis specifically refers to hormetic responses that originate in mitochondria and involve the activation of MSR pathways, including the MSR and UPRmt. The adaptive responses of the MSR and UPRmt are thought to function as forms of hormesis or mitohormesis in that they promote the activation of cellular repair mechanisms in response to mitochondrial stressors, which can ultimately lead to increased cellular stress resistance and improved health outcomes [44]. This process is thought to involve the activation of various protective pathways, such as induction of antioxidant defense systems, enhancement of mitochondrial biogenesis, and modulation of mitochondrial dynamics [8,45,46].
In severe cases of mitochondrial stress, the adaptive responses of the MSR and UPRmt may not be beneficial or hormetic. In fact, severe or chronic mitochondrial stress can lead to the accumulation of damaged or dysfunctional mitochondria, which can contribute to the development of a wide range of diseases, including metabolic disorders, neurodegenerative diseases, and cancer [47,48]. Mitochondrial dysfunction, characterized by impaired oxidative phosphorylation, has profound implications for several physiological processes. In the context of beta cell function, defective mitochondria disrupt energy production and subsequently impair insulin secretion, contributing to the pathogenesis of type 2 diabetes mellitus (T2DM) [49]. Furthermore, mitochondrial dysfunction in adipose tissues elicits chronic inflammation through increased ROS production and inflammatory signaling, thereby promoting metabolic disorders such as obesity and insulin resistance [50]. In the liver, impaired mitochondrial fatty acid oxidation results in lipid accumulation, leading to steatosis and progression to non-alcoholic steatohepatitis (NASH) [51]. Additionally, mitochondrial dysfunction-induced oxidative stress, inflammation, and cellular damage are implicated in the development of kidney and heart fibrosis, compromising organ function [52,53]. Furthermore, chronic or excessive activation of the MSR or UPRmt pathways may lead to the induction of pro-inflammatory pathways and the activation of cell death pathways, which can further exacerbate tissue damage and contribute to disease pathogenesis [54]. Therefore, while the adaptive responses of the MSR and UPRmt can function as forms of hormesis or mitohormesis under certain conditions, it is important to note that the optimal level of mitochondrial stress and the resulting adaptive responses may depend on the specific context and may differ between individuals or populations.
Multiple factors are involved in mediating the MSR and UPRmt. The Lon protease homolog (LONP1 or LON) is an ATPdependent serine protease in the mitochondrial matrix that plays a crucial role in mitochondrial protein quality control and stress response. It degrades misfolded, damaged, or unneeded proteins under normal conditions, but its activity increases under stressful conditions to prevent toxic protein aggregation [55]. If stress persists beyond LONP1’s capacity, it can lead to mitochondrial dysfunction and trigger a broader cellular stress response, which releases mitokines like fibroblast growth factor 21 (FGF21), growth differentiation factor 15 (GDF15), and humanin (HN) to coordinate a systemic response. Dysfunction in LONP1 or other MSR components is associated with diseases such as neurodegenerative diseases, cancers, and aging [56]. DAP3 binding cell death enhancer 1 (DELE1), an endonuclease found in the intermembrane space of mitochondria, is involved in the MSR and UPRmt. During stress, DELE1 is released into the cytosol and activates heme-regulated inhibitor (HRI), which reduces general protein synthesis while selectively increasing the translation of stress response factors. This pathway protects cells by reducing protein load and inducing the expression of protective factors, operating independently of the activating transcription factor 5 (ATF5) pathway. HRI, located in the cytosol, is activated by DELE1 during mitochondrial stress and phosphorylates eIF2α, decreasing protein synthesis and selectively enhancing translation of stress response factors (Fig. 1) [57]. This pathway is integral to the UPRmt, maintaining cellular homeostasis and restoring mitochondrial function in response to stress.

Regulation of fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15) expression by mitochondrial stress response (MSR) and mitochondrial unfolded protein response (UPRmt). The integrated stress response (ISR) is a cellular pathway activated in response to mitochondrial defects. In this pathway, a molecule called DAP3 binding cell death enhancer (DELE) plays a crucial role. DELE activates the heme-regulated inhibitor (HRI), which leads to the phosphorylation of eukaryotic initiation factor 2α (p-eIF2α) or triggers the ISR directly. This activation of the ISR facilitates the expression of key regulatory factors, such as activating transcription factors 4 (ATF4), ATF5, and C/EBP homologous protein (CHOP), which act as crucial regulators for the expression of FGF21 and GDF15. Additionally, an elevated ratio of adenosine monophosphate (AMP)/adenosine triphosphate (ATP), signaling energy stress, can induce the activation of AMP-activated protein kinase (AMPK). This activation occurs through a potential AKT serine/threonine kinase 1 (AKT1)-mediated mechanism. Ultimately, the activation of AMPK stimulates the expression of FGF21 or GDF15, further contributing to the regulatory processes involved in MSR and UPRmt. ETC, electron transport chain.
Dysfunction of mitochondrial pathways, including the MSR and UPRmt, has been implicated in the development of metabolic disorders such as insulin resistance and T2DM [58,59]. These pathways are also involved in the regulation of adipocyte differentiation and lipid metabolism, which are critical for maintaining metabolic homeostasis. Furthermore, dysregulated UPRmt signaling has been linked to the development of agerelated diseases, including neurodegenerative disorders and cancer [60,61]. UPRmt dysfunction can lead to the accumulation of damaged or misfolded proteins within mitochondria, which can trigger cell death pathways and contribute to disease development. The secretion of mitokines, which are induced by MSR and UPRmt activation, also plays a critical role in regulating systemic metabolism and influencing the development of metabolic and aging-related diseases [62]. Dysregulation of mitokine signaling has been linked to the development of a wide range of diseases, including obesity, insulin resistance, and neurodegenerative disorders [63,64].
Experimental studies have demonstrated that the MSR can be induced between multiple organ systems through various methods, such as partial inhibition of components of the electron transport system. In model organisms like Caenorhabditis elegans, mitochondrial perturbation in specific tissues (e.g., neurons) causes MSRs in distal tissues (e.g., the intestines) and leads to systemic effects, such as increased longevity [65]. These observations have led to the hypothesis that a cell non-autonomous signal is triggered upon sensing of mitochondrial stress and travels to other tissues in order to induce physiological responses. This elusive signaling molecule is termed a “mitokine” [65], and its identity and mechanisms of action are not fully understood. The concept of mitokines and their role in inter-tissue communication highlights the intricate interplay between various organ systems, and underscores the importance of maintaining mitochondrial function and communication for overall health and longevity [62]. Elucidating the molecular mechanisms underlying mitokine signaling and its downstream effects on systemic metabolism and physiological processes is critical for understanding the pathogenesis of metabolic and aging-related diseases, and developing new therapies to treat these conditions.
The discovery of mitokines in C. elegans has led to the identification of several relevant mitokines in mammals. Two of the most well-studied mitokines in mammals are FGF21 and GDF15. FGF21 is a hormone that is primarily secreted by the liver and adipose tissue in response to metabolic stress, such as fasting or exercise [66]. Recent studies have shown that FGF21 is also induced by mitochondrial stress and UPRmt activation, and that it can act as a mitokine to modulate systemic metabolism and promote cellular stress resistance [67]. GDF15 is a stress-induced cytokine that is secreted by a wide range of tissues, including skeletal muscle, adipose tissue, and the liver, in response to various types of stress, including inflammation and mitochondrial dysfunction [68-70]. GDF15 acts as a mitokine to modulate systemic metabolism and influence physiological processes such as appetite regulation, body weight, and glucose homeostasis [71,72]. Other potential mitokines identified in mammals include HN, mitochondrial open reading frame of the 12S rRNA-c (MOTS-c) [73], and FAM3 metabolism regulating signaling molecule C (FAM3C) [74]; however, the mechanisms of action and downstream effects of these signaling molecules are not fully understood.
Emerging evidence suggests that mitokines play important roles in normal physiology, in addition to their roles in cellular stress responses and inter-tissue communication. For example, FGF21 is involved in regulating energy metabolism and glucose homeostasis, and improves insulin sensitivity and protects against obesity and diabetes in animal models [75]. Similarly, GDF15 has been implicated in regulating appetite and body weight, and improves glucose tolerance and protects against obesity in animal models [72]. Furthermore, mitokines like FGF21 and GDF15 are induced by various metabolic stresses, such as fasting and exercise, and can modulate systemic metabolism and promote cellular stress resistance. These adaptive responses are thought to be critical for maintaining metabolic homeostasis and protecting against various types of cellular damage and stress. The identification of mitokines and their roles in normal physiology highlights the importance of maintaining mitochondrial function and communication for overall health and well-being, and underscores the potential of mitokine signaling as a therapeutic target for a wide range of metabolic and aging-related diseases [29]. Mitokines are believed to play important roles in a wide range of metabolic and agingrelated diseases, beyond mitochondrial diseases. Moreover, dysregulation of mitokine signaling has been implicated in the development of various age-related diseases, such as Alzheimer’s disease, Parkinson’s disease, and sarcopenia [62,76,77]. The accumulation of mitochondrial damage and dysfunction that occurs during aging can lead to activation of the MSR and UPRmt pathways, which can in turn induce the secretion of mitokines and other signaling molecules that contribute to disease pathogenesis.
MITOKINES
Mitokines are signaling molecules, encoded by both nuclear (e.g., GDF15 and FGF21) and mitochondrial DNA (e.g., HN), that are secreted in response to mitochondrial stress or the UPRmt, facilitating inter-organ or inter-tissue communication to manage cellular responses to mitochondrial distress. The discovery of mitokines originated from studies conducted in worms, with the FMRFamide-like neuropeptides 2 (FLP-2) and serotonin being among the first signaling molecules identified to function in the initiation of inter-organ mitochondrial stress-related signaling in C. elegans [78,79]. However, these signaling molecules are insufficient to induce MSRs in distant tissues. It was later discovered that the Wnt/EGL-20 ligand acts as a mitokine in C. elegans, inducing adaptive responses in response to mitochondrial stress [80].
In mammals, FGF21 and GDF15 are the most extensively studied mitokines, with other candidate mitokines such as HN, MOTS-c, and angiopoietin-like 6 (ANGPTL6) being proposed [81-83]. Many more molecules likely function as mitokines, and their discovery and characterization will broaden our understanding of mitochondrial communication and its role in health and disease.
Mitokines have a variety of intracellular, autocrine, paracrine, and endocrine functions [84]. In this review, we focus on the endocrine actions of mitokines related to mitochondrial stress and adaptations. It is important to note that mitokines often have additional functions independent of their role in mitochondrial stress signaling, and distinguishing between these functions is crucial to avoid ambiguity and to clarify their specific actions. In this context, we first highlight the broad functions of FGF21 and GDF15, and then delve into their specific mitokine actions in regulating inter-tissue communication and metabolic homeostasis.
MITOKINE ACTION OF FGF21
As described earlier, several studies have provided evidence that FGF21 is upregulated in response to mitochondrial stress and during the UPRmt [63,85-88]. The molecular and cellular mechanisms underlying FGF21 induction during mitochondrial stress or the UPRmt are not fully understood. FGF21 was found to be upregulated via the integrated stress response (ISR) in mice with impaired autophagy in skeletal muscle. This study showed that activation of the ISR by mitochondrial stress led to the induction of activating transcription factor 4 (ATF4), which in turn increased FGF21 expression (Fig. 1) [63]. ATF5 plays a role in the response to mitochondrial stress and may be responsible for the induction of FGF21 (Fig. 1) [89]. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is a key transcriptional coactivator involved in the regulation of mitochondrial biogenesis and function. Several studies have suggested that PGC-1α may play a role in FGF21 induction during mitochondrial stress. For example, a study by Khan et al. [90] showed that FGF21 induction in response to mitochondrial dysfunction was associated with increased PGC-1α expression in skeletal muscle. It has been suggested that adenosine monophosphate-activated protein kinase (AMPK) might be involved in FGF21 induction during mitochondrial stress. For instance, a study by Salminen et al. [91] showed that activation of AMPK by mitochondrial stress led to increased FGF21 expression in adipose tissue (Fig. 1).
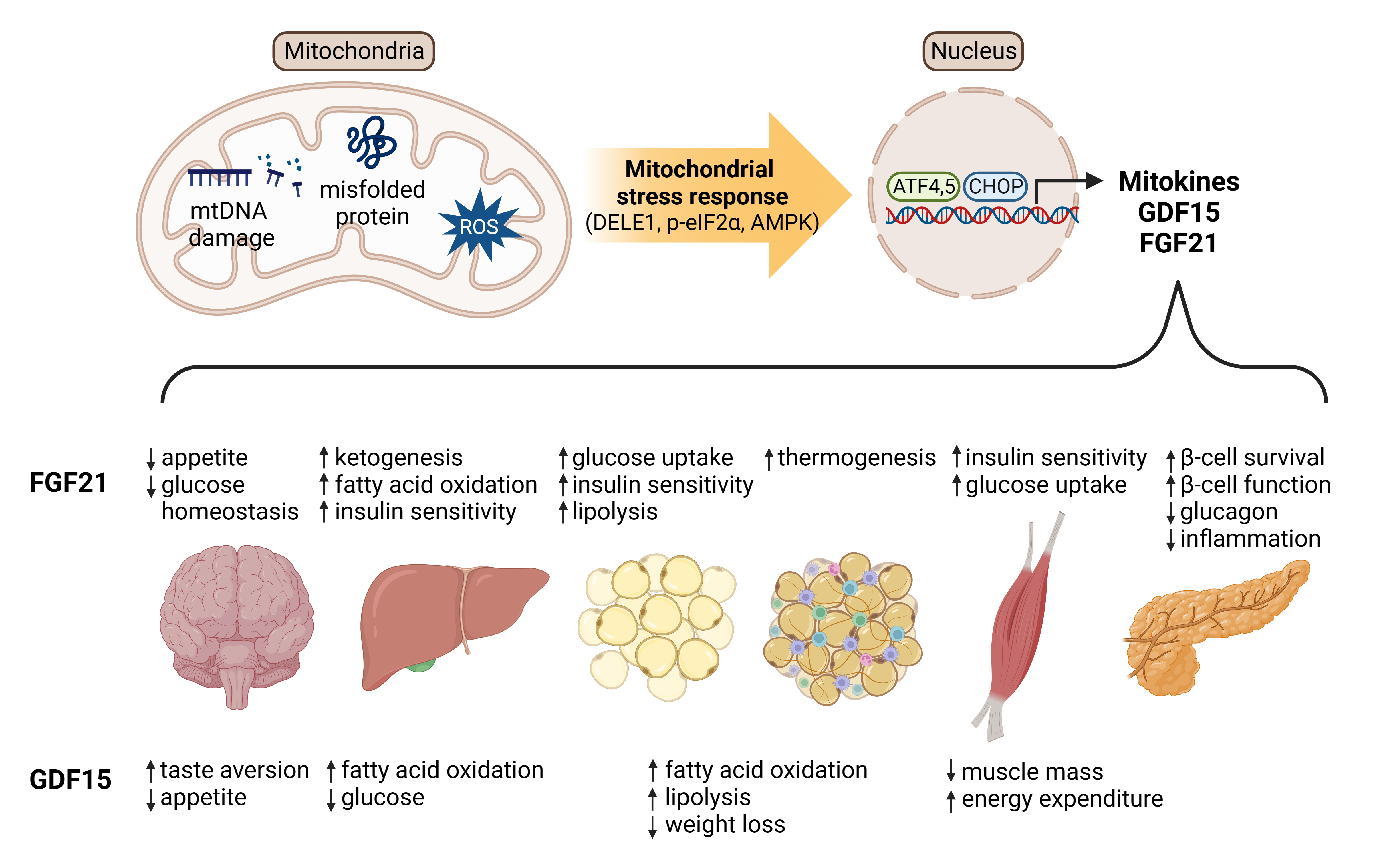
FGF21 is a hormone that plays a significant role in regulating metabolism, energy homeostasis, and mitochondrial function. Its influence on mitochondria has been observed in various tissues, such as skeletal muscles, adipose tissue, and the brain. FGF21 has been implicated in promotion of mitochondrial biogenesis, particularly in skeletal muscles. It activates the PGC-1α pathway, which is a key regulator of mitochondrial biogenesis and function [92,93]. FGF21 may partially regulate mitophagy, a process that selectively degrades damaged or dysfunctional mitochondria to maintain mitochondrial quality and function [76]. FGF21 has been linked to the regulation of mitochondrial dynamics, which involves the balance between mitochondrial fusion and fission processes that affect mitochondrial morphology, distribution, and function [94]. In adipose tissue, FGF21 improves mitochondrial oxidative function through the activation of AMPK and sirtuin 1 [95]. The overall metabolic and thermogenic effects of FGF21 may indirectly regulate mitochondrial function in various brain regions. In specific target areas of FGF21 in the brain, mitochondria are likely more directly modulated by FGF receptor-mediated intracellular signaling. The brain plays an important role in mediating some of the actions of FGF21. The receptor-mediated role of FGF21 affects the hypothalamus, a key brain region involved in controlling energy balance, food intake, and body weight (Fig. 2) [96]. By binding to its receptors fibroblast growth factor receptor 1c (FGFR1c) and β-Klotho in the hypothalamus, FGF21 can modulate appetite and energy expenditure, leading to weight loss [97]. FGF21 has been reported to exert neuroprotective effects in various neurological disorders, such as Alzheimer’s disease and Parkinson’s disease, through its receptors FGFR1c and β-Klotho, which activate intracellular signaling pathways that promote neuronal survival and reduce neuroinflammation [98,99]. Corticotropin-releasing factor (CRF), which is a key component of the brain’s stress response system, is regulated by FGF21 [100]. This regulation may be associated with mitochondrial alterations because CRF modulates mitochondria in neurons via nuclear factor κB and the mitochondrial fission factor dynamin-1-like protein [101]. The receptor-mediated action of FGF21 in the brain plays a central role in glucose homeostasis. It has been suggested that FGF21 can modulate glucose metabolism by acting on proopiomelanocortin (POMC), agouti-related protein/neuropeptide Y (AgRP/NPY), steroidogenic factor 1 (SF1) neurons in the hypothalamus and other brain regions (Fig. 2) [102,103].

The pathophysiological roles of fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15) in the metabolic disease. FGF21 regulates appetite, enhances ketogenesis, improves insulin sensitivity, promotes lipolysis, and influences glucose homeostasis. GDF15 regulates appetite, leads to weight loss, decreases muscle mass, promotes lipolysis and fatty acid oxidation, and affects liver metabolism.
The circulating half-life of FGF21 in humans is relatively short, with a range of 1 to 2 hours, primarily due to its rapid degradation by proteases, including a key endopeptidase known as fibroblast activation protein (FAP) [104,105]. FAP specifically cleaves FGF21 at a certain site, rendering it inactive and thus diminishing its biological activity. Interestingly, this degradation mechanism does not apply to mice as FAP does not cleave FGF21 in them. While the synthesis, secretion, receptor binding, and elimination of FGF21 are largely conserved between humans and mice, this difference in the role of FAP results in a divergence in FGF21 metabolism and plasma degradation between the two species [105]. However, FGF21 is not degraded by FAP in mice. This difference leads to a difference in FGF21 metabolism between the two species. Consequently, FGF21 might have a longer circulating half-life in mice than in humans. This distinction in plasma degradation of FGF21 between humans and mice is an important factor to consider when translating findings from mouse models to human physiology and when developing FGF21-based therapeutics.
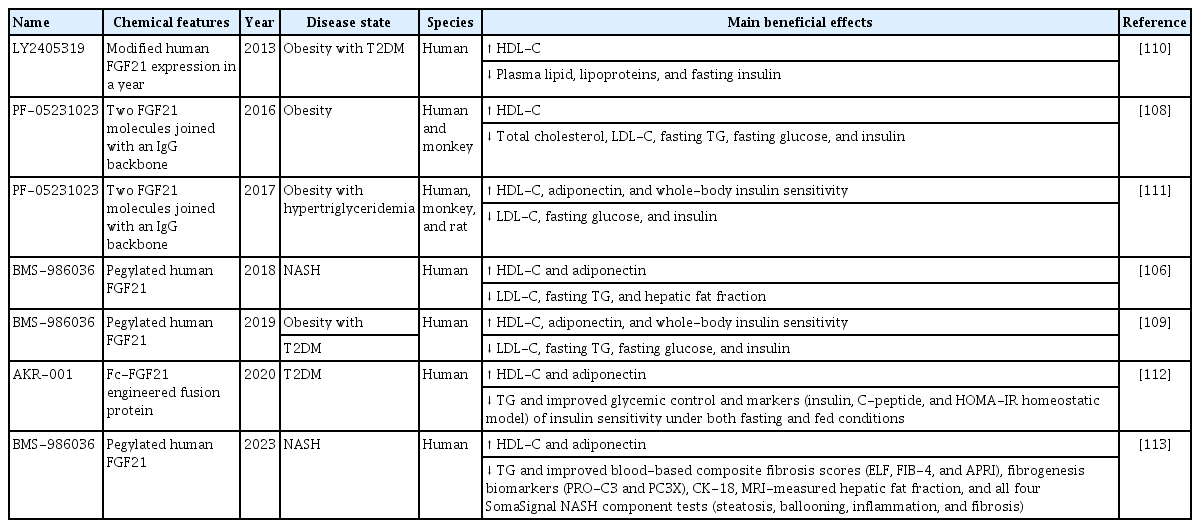
To date, six randomized clinical trials have been conducted testing the therapeutic potential of four human FGF21 analogs or mimetics in T2DM or obesity, with or without another defined metabolic disorder such as fatty liver disease (Table 1). A PEGylated FGF21 analog, Pegbelfermin (BMS-986036), was developed by Bristol-Myers Squibb (New York, NY, USA) for the treatment of NASH. Phase II clinical trials showed improvements in liver fat content and liver fibrosis in patients with NASH [106]. An FGF21 analog, efruxifermin (EFX, formerly known as AKR-001), was developed by Akero Therapeutics (South San Francisco, CA, USA) for the treatment of NASH. A phase IIa clinical trial (the BALANCED study) of EFX was completed, which demonstrated improvements in liver fat content, liver enzymes, and other metabolic parameters in patients with NASH [107]. A long-acting FGF21 analog, PF-05231023, was developed by Pfizer (New York, NY, USA) for the treatment of T2DM and obesity. Phase I clinical trials showed improvements in lipid profiles and body weight in patients with T2DM [108]. However, in a phase II trial, the treatment led to an increase in adverse events related to bone mineral density, resulting in discontinuation of the trial (Table 1) [106,108-113].
FGF21 clinical trials
Several small molecules have been identified that can modulate mitochondrial function and in turn impact FGF21 expression. Metformin is a widely used antidiabetic drug that activates AMPK and inhibits complex I of the mitochondrial electron transport chain. Metformin increases FGF21 expression in various experimental settings, potentially through its effects on mitochondrial function [114]. Thiazolidinediones (TZDs), such as rosiglitazone and pioglitazone, are peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists used to treat T2DM. PPAR-γ coactivates PGC-1α, which is involved in the regulation of FGF21 expression. TZDs increase FGF21 expression in vitro and in vivo, potentially through their effects on PPAR-γ and PGC-1α [115]. The 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) is an AMPK activator that modulates mitochondrial function. AICAR increases FGF21 expression in skeletal muscle cells, potentially through its effects on AMPK activation and mitochondrial function [92]. The pharmacological efficacy of some small molecule pharmaceuticals could be related to FGF21 induction. However, it is essential to note that these small molecule pharmaceuticals have multiple targets and mechanisms of action. While FGF21 induction might contribute to their pharmacological efficacy, it is not the sole determinant of their therapeutic effects. In many cases, the relationship between FGF21 induction and the pharmacological efficacy of small molecule pharmaceuticals remains to be fully elucidated.
MITOKINE ACTION OF GDF15
Recent clinical observation studies have shown that GDF15 levels are elevated in patients with primary mitochondrial diseases (PMDs), such as those caused by mutations in mitochondrial DNA or nuclear genes affecting mitochondrial function [116,117]. GDF15 levels were correlated with PMD severity and could be used as a reliable index of disease progression [118-120]. In 2017, Chung et al. [54] reported that circulating GDF15 levels were upregulated in response to mitochondrial dysfunction in mouse models. The increase in GDF15 levels was dependent on mitochondrial stress, leading to changes in systemic metabolism. In 2019, Mottis et al. [121] and Tezze et al. [122] further supported the classification of GDF15 as a mitokine due to its role in modulating systemic metabolism and its upregulation in response to mitochondrial stress. These animal studies helped to establish GDF15 not only as a potential biomarker for mitochondrial disorders but also as a molecule with a functional role in mediating the body’s response to mitochondrial dysfunction.
GDF15, also known as macrophage inhibitory cytokine-1 (MIC-1), is a protein encoded by the GDF15 gene in humans. It is a member of the transforming growth factor-β superfamily. Under normal physiological conditions, GDF15 is expressed at low levels in most organs. However, its expression can significantly increase in response to tissue injury or stress in various organs, such as the liver, kidneys, heart, and lungs. GDF15 levels can fluctuate under various physiological and pathophysiological conditions, often related to mitochondrial stress (recently reviewed by Johann et al. [123]). For instance, intense exercise can result in significant release of GDF15 from skeletal muscle. Additionally, factors such as aging, injury, inflammation, and numerous diseases, particularly cancers, are associated with elevated GDF15 serum levels [124].
GDF15 expression is related to the UPRmt and ISR [125,126]. When mitochondrial stress occurs, the UPRmt activates transcription of GDF15, which in turn is modulated by the ISR. The precise molecular mechanisms by which the ISR and UPRmt integrate to express GDF15 are not fully understood. UPRmt activation leads to the upregulation of various nuclear-encoded mitochondrial chaperones and proteases, which help to maintain mitochondrial proteostasis [125]. On the other hand, the ISR is activated in response to various cellular stress conditions, including endoplasmic reticulum stress, amino acid starvation, and oxidative stress, and aims to restore cellular homeostasis by modulating protein synthesis, folding, and degradation [126]. One possible connection between the ISR and UPRmt in the context of GDF15 expression involves ATF4 (Fig. 1). When the ISR is activated in response to cellular stress, translation of ATF4, a transcription factor, is upregulated. ATF4 can then translocate to the nucleus and induce the transcription of its target genes, which are involved in the stress response, amino acid metabolism, and redox homeostasis. Under mitochondrial stress, ATF4 has been implicated in the regulation of GDF15 expression [75,127,128]. In addition, ATF5 and C/EBP homologous protein (CHOP) are two transcription factors involved in GDF15 expression. The regulation of GDF15 expression by ATF4, ATF5, and CHOP through the UPRmt may be cell- or stress-specific (Fig. 1). The cellular context and type of stress experienced by cells can influence the activation of transcription factors and expression of GDF15 [31,89]. While deletion of ATF4, ATF5, and CHOP might impair certain aspects of the UPRmt and regulation of GDF15 expression, it is unlikely to completely abrogate the UPRmt or the mitokine action of GDF15. Other factors and pathways in the UPRmt and alternative regulatory mechanisms may still contribute to the overall stress response and GDF15 function.
Four research groups simultaneously identified GDNF family receptor alpha like (GFRAL) as the receptor for GDF15, which signals through the co-receptor RET [129-132]. All four groups found that GDF15 binds specifically to GFRAL, which is expressed on the surface of a subset of neurons in the area postrema and nucleus tractus solitarius in the brainstem. Binding of GDF15 to GFRAL leads to recruitment and activation of the co-receptor RET, which activates downstream signaling pathways involved in the regulation of appetite and energy homeostasis (Fig. 2). All four groups found that GFRAL signaling plays a crucial role in the regulation of feeding behavior and body weight. Mice lacking GFRAL or GDF15 exhibit increased food intake and body weight gain, while administration of GDF15 reduces food intake and leads to weight loss. Recently, a study discovered that membrane-bound matrix metalloproteinase 14 (MT1-MMP/MMP14) negatively regulates GFRAL, the cognate receptor of GDF15, in the context of obesity. Genetic ablation of MT1-MMP in GFRAL+ neurons restored GFRAL expression, leading to reduced weight gain and food intake in obese mice, while GFRAL depletion negated these anti-obesity effects. These findings suggest that targeting negative regulators of the GDF15-GFRAL pathway, such as MT1-MMP, could be a therapeutic strategy for obesity [133]. In summary, GFRAL signaling has been suggested to be a potential therapeutic target for obesity and cachexia (muscle wasting) because its activation leads to reduced food intake and body weight loss (Fig. 2).
FGF21 and GDF15 play a role in mediating the MSR. Although they have some overlapping functions, such as improving insulin sensitivity, these proteins also elicit unique effects on various physiological processes. A study by Kang et al. [75] published in 2021 using mice with hepatic mitoribosomal defects demonstrated that FGF21 specifically ameliorated glucose clearance, energy expenditure, and thermogenesis. On the other hand, GDF15 was beneficial for fat mass and hepatic steatosis. Adding to the complexity, GDF15 has differential effects depending on its mode of induction. A study by Klein et al. [71] published in 2021 reported that pharmacological administration of systemic GDF15 led to suppression of appetite and running activity in mice. However, when GDF15 levels were naturally elevated by running, which causes mitochondrial stress, these effects were not observed. These findings suggest that FGF21 and GDF15 have distinct roles in regulating various physiological processes and that their effects may depend on the context in which they are induced or administered. They also suggest that exercise-induced GDF15 and pharmacological GDF15 may have different effects on systemic metabolism. Exercise-induced GDF15, which is naturally elevated due to mitochondrial stress caused by physical activity, does not appear to suppress appetite or running activity in contrast with pharmacological GDF15. This difference in effects could be attributed to the context in which GDF15 is induced, as well as possible differences in the level or duration of GDF15 elevation.
Based on the current understanding of GDF15 and its role in metabolism and energy homeostasis, it is reasonable to consider GDF15 or its modified analogs as potential therapeutics for obesity and related metabolic disorders (Table 2). Recombinant GDF15 and GFRAL pathway proteins and antibodies were tested in a variety of disease models. Fc-fused GDF15 molecules with longer half-lives than GDF15 were synthesized and tested in obese mice, rats, and monkeys [134]. These molecules demonstrated strong efficacy in lowering body weight in every species tested and improved multiple metabolic parameters. A glucagon-like peptide-1 (GLP-1)/GDF15 fusion protein engineered via fusing GLP-1 and an GDF15 analog via a peptide linker and conjugating it to a fatty acid showed superior potency in vitro and improved metabolic parameters in mice and cynomolgus monkeys [135]. One research group from a pharmaceutical company developed the GDF15 analog LY3463251, which is a potent agonist of the GFRAL/RET receptor with prolonged pharmacokinetics (Table 2) [136]. Interestingly, a 12-week multiple ascending dose study with overweight and obese participants showed that LY3463251 induced significant decreases in food intake and appetite scores associated with modest body weight reduction independent of nausea and emesis [136]. In this clinical trial, LY3463251-induced nausea and emesis were dose-dependent and common at the highest dose tested, including upon single- and multiple-dose exposures. This is consistent with prior clinical evidence suggesting that GDF15 has adverse effects in humans [137,138].
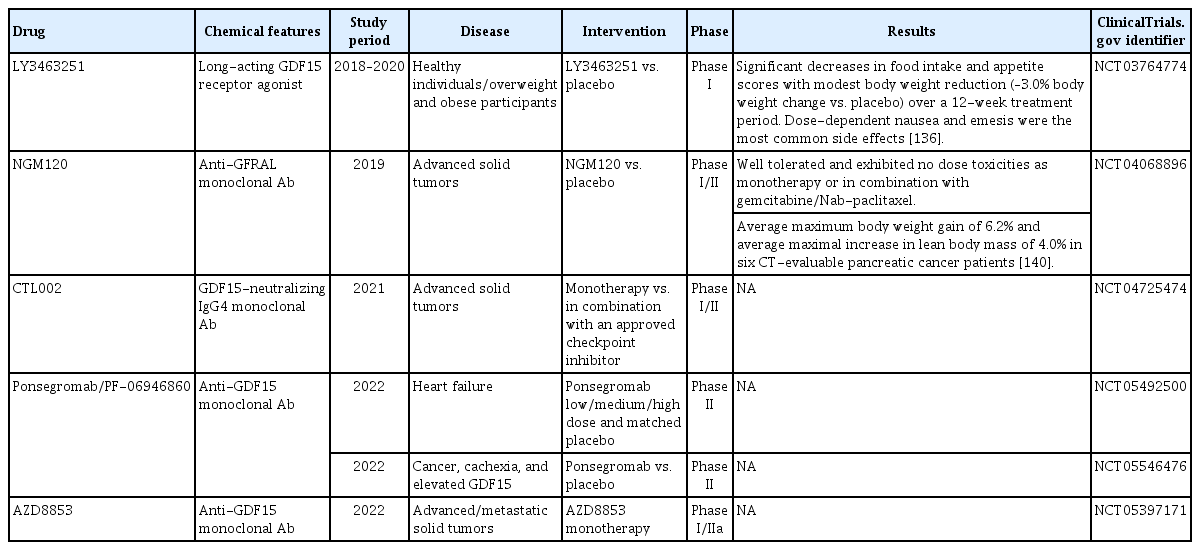
GDF15 and GFRAL clinical trials
On the other hand, GDF15 neutralization and blockade of GFRAL action may have beneficial effects in certain contexts, particularly in situations where elevated levels of GDF15 contribute to nausea, anorexia, and wasting syndrome, including cancer-associated cachexia (Table 2). A recent study showed that cachectic mice treated with an anti-GDF15 antibody (mAB2) exhibited body weight gain, muscle mass restoration, and improved muscle function and physical performance. The improvements were primarily attributed to increased caloric intake, while altered gene expression in cachectic muscles was restored in both caloric intake-dependent and -independent manners [139]. The study found that GDF15 levels were elevated in cancer patients receiving platinum-based chemotherapy and were linked to weight loss in colorectal cancer patients. GDF15-knockout mice exhibited reduced anorexia and weight loss, while GDF15 neutralization using the monoclonal antibody mAB1 improved survival and alleviated side effects in both mice and nonhuman primates. Three anti-GDF15 monoclonal antibodies and one anti-GFRAL monoclonal antibody are in phase I/II clinical trials for the prevention and treatment of cachexia in advanced cancer patients (Table 2) [136,140]. In addition, ponsegromab, an anti-GDF15 monoclonal antibody, is in a phase II clinical trial evaluating the frequency, severity, and burden of symptoms as well as physical limitations in participants with heart failure (NCT05492500). The findings of preclinical studies suggest that GDF15 neutralization is a potential therapeutic approach for mitigating chemotherapy-induced side effects and enhancing patients’ quality of life, and ongoing clinical trials will show its applicability in humans [138].
MITOCHONDRIA-DERIVED PEPTIDES ACTING AS MITOKINES
Mitochondria-derived peptides (MDPs) are an emerging class of microproteins, which are biologically active peptides composed of fewer than 100 amino acids [141,142]. These peptides are encoded by small open reading frames within mitochondrial DNA [143]. To date, eight MDPs have been identified, with HN and MOTS-c being the most extensively studied exercise-inducible mitokines. HN and MOTS-c are well-known mitokines that play essential roles in cellular homeostasis, cytoprotection, and metabolic regulation. The small HN-like peptides (SHLPs) 1–6 are a more recently discovered class of MDPs that share functional similarities with HN [143,144]. These peptides exhibit cytoprotective effects, attenuating oxidative stress, enhancing mitochondrial efficiency, and promoting systemic glucose metabolism [144]. Due to these overlapping functions, SHLPs are believed to contribute to the beneficial effects of MDPs. Despite the growing understanding of these MDPs, many additional uncharacterized MDPs are hypothesized to exist [145]. Some of these undiscovered peptides may also qualify as mitokines, potentially expanding our understanding of mitochondrial signaling and regulation. However, characterization of these novel MDPs is technically challenging due to their small size, low abundance, and unique sequence features. Recent advances in molecular biology and analytical techniques have made the identification and characterization of these elusive MDPs more feasible [143]. Low-grade mitochondrial stress, known as mitohormesis, can promote health and longevity. In this context, partial Crif1 deficiency in hypothalamic POMC neurons increases expression of β-endorphin and mitochondrial DNA-encoded MOTS-c, leading to high-turnover metabolism and resistance to obesity [146].
HN is a 24-amino acid polypeptide encoded by the mitochondrial 16s rRNA gene and is expressed in various tissues such as the heart, brain, liver, colon, skeletal muscle, kidneys, testes, and vascular wall. It is a suppressor of apoptosis that promotes cell survival under various stress conditions [147]. One mechanism via which HN exerts anti-apoptotic effects is by binding to cell surface receptors, which subsequently upregulate the phosphoinositide 3-kinase/AKT signaling pathway, a well-known mediator of cell survival and growth. Additionally, HN has been reported to enhance mitochondrial respiration and biogenesis, suppress calcium overload, inhibit the Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways, and reduce ROS production and oxidative stress [148]. These actions collectively contribute to the cytoprotective role of HN. MOTS-c is another microprotein with the potential to regulate cell metabolism and is thus an attractive candidate as an exercise mimetic. MOTS-c is regulated by mitochondrial stress and can inhibit the folate cycle, leading to decreased purine biosynthesis, particularly in muscle tissue [73]. This inhibition activates AMPK, which protects against insulin resistance and promotes metabolic homeostasis. In situations of metabolic stress, such as glucose restriction, MOTS-c translocates to the nucleus, where it contributes to the cellular stress response by modulating the expression of various genes, including those involved in antioxidant defense mechanisms [149].
The sensitivity of mitokines like HN and MOTS-c to physical exercise suggests that their health-promoting potentials can be harnessed through lifestyle interventions, such as regular physical activity. By understanding the molecular mechanisms underlying the actions of these mitokines, researchers can develop strategies to exploit their beneficial effects on cellular metabolism, the stress response, and overall health. In the following section, we will outline how such lifestyle interventions can impact the function of mitokines and contribute to improved health outcomes.
CONCLUSIONS
Mitokines are signaling molecules released by mitochondria that play a crucial role in cellular communication, energy metabolism, and overall health. Despite advancements in understanding mitokines, there are still unclear aspects that require further investigation, including the signaling pathways responsible for trans-tissue communication and the optimal levels of mitokines for promoting health. It is important to study potential side effects and risks of manipulating mitokine levels to ensure safe and effective therapies. Exploring inter-mitochondrial signaling in response to stress could reveal new mechanisms and molecules as potential mitokines, expanding our understanding of their roles in age-related diseases. Modulating mitokine signaling in response to exercise and pharmaceutical agents offers opportunities for personalized interventions and the development of novel therapies for various conditions. This research will reshape our understanding of mitochondria’s functions in overall health and disease, leading to targeted therapies and improved quality of life for individuals affected by age-related and chronic diseases.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2023R1A2C3003438) and Global Research Laboratory (GRL) Program through the National Research Foundation of Korea(NRF) funded by the Ministry of Science and ICT (2017K1A1A2013124).
Acknowledgements
The authors would like to acknowledge the members of Dr. Minho Shong’s research group at the Chungnam National University for their contributions to the field of mitochondrial stress and mitokines, and for their insights and guidance during the preparation of this review article.