- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 34(1); 2010 > Article
-
ReviewThe Incretins and Pancreatic β-Cells: Use of Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Polypeptide to Cure Type 2 Diabetes Mellitus
- Mi-Hyun Kim1, Moon-Kyu Lee2
-
Korean Diabetes Journal 2010;34(1):2-9.
DOI: https://doi.org/10.4093/kdj.2010.34.1.2
Published online: February 28, 2010
- 4,169 Views
- 39 Download
- 9 Crossref
1Division of Endocrinology and Metabolism, Samsung Biomedical Research Institute (SBRI), Seoul, Korea.
2Division of Endocrinology and Metabolism, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- Corresponding author: Moon-Kyu Lee. Division of Endocrinology and Metabolism, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 50 Ilwon-dong, Kangnam-gu, Seoul 135-710, Korea. leemk@skku.edu
Copyright © 2010 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Type 2 diabetes mellitus (T2DM) is increasing in prevalence worldwide. The complications associated with T2DM result in increased mortality and financial cost for those affected. T2DM has long been known to be associated with insulin resistance in peripheral tissues and a relative degree of insulin deficiency. However, the concept that insulin secretion and insulin sensitivity are not linked through a hyperbolic relationship in T2DM has continuously been demonstrated in many clinical trials. Thus, in order to prevent and treat T2DM, it is necessary to identify the substance(s) that will improve the function and survival of the pancreatic β-cells in both normal and pathologic conditions, so that production and secretion of insulin can be enhanced. Incretin hormones, such as glucagon-like peptide (GLP)-1 and glucose-dependent insulinotropic polypeptide (GIP), have been shown to lower the postprandial and fasting glucose and the glycated hemoglobin levels, suppress the elevated glucagon level, and stimulate glucose-dependent insulin synthesis and secretion. In this report, we will review the biological actions and mechanisms associated with the actions of incretin hormones, GLP-1 and GIP, on β-cell health and compare the differences between GLP-1 and GIP.
- The prevalence of type 2 diabetes mellitus (T2DM) is increasing worldwide. The World Health Organization (WHO) estimates that approximately 180 million individuals are currently affected by T2DM, with the number expected to double by the year 2030 [1]. The situation seems to be more serious in Asian countries, such as India and China, which currently have the greatest number of patients with diabetes mellitus [2]. Compared to Western countries such as the United States, Asian countries tend to have more patients with diabetes in the younger generations and the difference in prevalence between urban and rural community has been gradually declining [3]. The financial and societal costs attributable to T2DM are substantial. In the U.S. alone, the estimated total costs were $132 billion in the year 2002 [4].
- Recently, β-cell dysfunction and the substantial reduction in the maximum capacity to secret insulin were demonstrated to be the primary metabolic defects in patients with T2DM [5]. Indeed, according to the United Kingdom Prospective Diabetes Study (UKPDS), β-cell function is generally diminished by more than 50% by the time an individual is diagnosed with T2DM [4]. Many individuals who are insulin resistant never develop T2DM, as long as they have normal β-cell function that is competent enough to adapt to the state of the insulin resistance [6]. Thus, the deterioration of β-cell function mainly contributes to the difficulty in maintaining adequate glycemic control in patients with T2DM. In particular, the degree of β-cell dysfunction seems to be greater in Asians than in individuals from Western countries. In Korean subjects, the impaired insulin secretion could be demonstrated in those with impaired glucose tolerance or impaired fasting glucose, and β-cell dysfunction is the only parameter associated with the development of T2DM in a prospective cohort study, and the selective β-cell loss was noticed in pancreatic islets of the patients with T2DM in a postmortem study [7-9]. Japanese people with normal glucose tolerance also have an insulin secretory capacity that is insufficient to compensate for the obesity-induced insulin resistance [10]. The widely-used anti-diabetic agents, such as metformin, sulfonylureas, and thiazolidinediones, attenuate the pathophysiology of T2DM mainly through the promotion of insulin sensitivity and/or insulin secretion, but none of these are likely to prevent T2DM via the improvement of β-cell dysfunction. Thus, there is an increasing need for new agents to improve β-cell dysfunction.
- Incretins, glucagons-like peptide (GLP)-1 and glucose-dependent insulinotropic polypeptide (GIP), are hormones secreted from the gut in response to nutrient entry. They play a major role in glucose homeostasis via the insulinotropic mechanism and the proliferative and anti-apoptotic mechanisms in β-cells [11]. In many individuals with T2DM or insulin resistance, it has been demonstrated that the GLP-1 level is decreased and GIP response to increase insulin secretion is impaired [12,13]. Additionally, genetic variation in GIPR, even in non-diabetic individuals, was recently shown to be associated with a diminished insulin response and incretin effects in response to an oral glucose challenge [14]. However, the intact GLP-1 level was not decreased in Japanese patients with T2DM [15]. It is still not firmly established whether incretins are really associated with the pathogenesis of T2DM and whether they exert beneficial effects on insulin secretion and β-cell dysfunction or β-cell mass in subjects with T2DM.
INTRODUCTION
- In 1960, extracts of mucosa from the porcine upper small intestine were used as a treatment for diabetes [16]. In 1932, the effect of the unidentified substance was referred to as the "incretin effect" [17]. Some years later, a polypeptide was discovered and named "gastric inhibitory polypeptide (GIP)" due to its inhibitory effect on gastric acid secretion in dogs. GIP was later renamed "glucose-dependent insulinotropic polypeptide (GIP)", because its gastric inhibitory effect was found to be weak [18]. Although GIP was shown to be a potent stimulator of insulin secretion, removal of GIP from gut extracts via immunoadsorption did not eliminate the incretin effect, providing evidence for the existence of an additional peptide, later called GLP-1, with incretin-like activity [19]. In 1983, the gene encoding proglucagon, the precursor of glucagon, was found to include the sequence of two peptides (GLP-1 and GLP-2), in addition to glucagon itself. Proglucagon is expressed in both the pancreatic a-cells and the intestinal L-cells and its primary transcripts and translation products are identical in the two types of cells. However, the post-translational processing differs markedly in these two tissues [20,21].
- GLP-1 and GIP are made from proglucagon and proGIP protein precursors in the intestine or pancreas. First, bioactive GLP-1 is generated from proglucagon through cleavage of single arginine residue that flank GLP-1 by the intestine-specific prohormone convertase (PC) 1 or 3 [22]. Second, bioactive GIP is generated from proGIP via cleavage by PC 1 or 3 of single arginine residue that flanks GIP [11]. GLP-1 is secreted from L-cells, which are located mainly in the distal ileum and colon. In contrast, GIP is released form K-cells that are localized to a more proximal region (duodenum and jejunum). These cells are in direct contact with luminal nutrients and neural and vascular tissues, which could regulate GLP-1 and GIP secretion by a variety of mechanisms including endocrine factors [23]. As soon as GLP-1 and GIP are secreted, they are inactivated by the ubiquitous proteolytic enzyme dipeptidyl peptidase-4 (DPP-4), which is a serine protease that specifically cleaves dipeptides from the protein that contains an alanine or proline residue in their N-terminal sequences. Thus, bioactivities of GLP-1 and GIP are maintained for very short periods of 2 min and 7 min, respectively [24].
METABOLISM OF GLP-1 AND GIP
- The biological actions of GLP-1 and GIP in β-cells are mainly undertaken through their receptors, glucagon-like peptide-1 receptor (GLP-1R) and glucose-dependent insulinotropic polypeptide receptor (GIPR), respectively. GLP-1R and GIPR belong to the class B family of 7 transmembrane-spanning, heterotrimetric G-protein-coupled receptors [25,26], and are expressed in α-, β-, and δ-cells of the pancreatic islets. The binding of GLP-1 and GIP to the N-terminal extracellular regions of their receptors activates the G protein and its downstream signaling pathways, which result in i) the exocytosis of insulin storage granules, ii) the stimulation of insulin synthesis, iii) differentiation of pancreatic exocrine cells toward a more endocrine-like phenotype, iv) expansion of β-cell mass, and v) the reduction of endoplasmic reticulum (ER) stress (Table 1).
- First, the insulin granule exocytosis is likely to result from GLP-1-induced KATP channel closure and adenylyl cyclase activation, in which the former induces membrane depolarization and opens a voltage-dependent Ca2+ channel, and subsequently increases the influx of the extracellular Ca2+ into the cell, and the latter increases intracellular cAMP levels and consequently activates Epac2, which releases Ca2+ from the ER. The increased [Ca2+]i leads to the exocytosis of insulin storage granules out of β-cells [11].
- Second, GLP-1-stimulated insulin synthesis appears to be responsible for cAMP/PKA- or cAMP/Epac/ TORC2 (a CREB coactivator)-dependent signaling, which phosphorylates the cAMP response element binding protein (CREB) [27] and insulin receptor substrate (IRS)-2 and subsequently increases the expression of pancreas/duodenum homeobox-1 (Pdx-1) and the binding of Pdx-1 to the insulin gene promoter [28]. The importance of Pdx-1 in GLP-1-stimulated insulin synthesis was confirmed by the evidence that the GLP-1 agonist exendin-4 failed to increase the plasma insulin level, pancreatic insulin content, and insulin mRNA expression in β-cellPdx-/- mice compared to the wild type mice [29].
- Third, the differentiation of pancreatic exocrine cells toward β-cells may be attributable to GLP-1-stimulated Pdx-1 gene transcription [30]. Indeed, GLP-1 increased the expression of β-cell-specific genes such as insulin, glucose transporter 2 (GLUT2), and glucokinase in human and rat pancreatic ductal cells transfected with Pdx-1 compared with those transfected with null vector [30].
- Fourth, β-cell expansion may result from the inhibition of FoxO1 and activation of Pdx-1 via cAMP/PKA/CREB/IRS-2 signaling or PI3K/AKT signaling or cell cycle regulator cyclin D1 up-regulation through MAPK signaling. In INS-1 cells transduced with constitutively active nuclear-FoxO1, the anti-apoptotic effect of GLP-1R agonist was abolished with extrusion of anti-apoptotic protein Pdx-1 into the cytoplasm [31].
- Further, the reduction of ER stress is likely to result from GADD34-induced dephosphorylation of eIF2α. In an Ins-1 β-cell line, ER stress was induced by thapsigargin, a non-competitive inhibitor of SERCA, and GLP-1R agonist potentiated the translation of ATF4 and the expression of its target gene GADD34, thereby increasing GADD34-mediated dephosphorylation of eIF2α [32].
- Like GLP-1, GIP shows similar biological activity (the insulinotropic and proliferative effects) on pancreatic β-cells and has many common steps of intracellular signal transduction, such as membrane potential change, intracellular calcium response, and cAMP response. However, several different biological actions have also been demonstrated for GIP and GLP-1 on β-cells (Table 1).
- First, a novel role of GIP in the regulation of KV channel was identified as a potential mechanism whereby GIP modulates insulin secretion. In INS-1 cells, GIP reduced an A-type peak ionic current amplitude of Kv1.4. The mutant form of Kv1.4 with Thr/Ala substitutions in a PKA-phosphorylated site reversed the GIP-reduced Kv1.4 peak current amplitude and subsequently reduced glucose-dependent insulin secretion [33]. In addition, GIP did not regulate fasting glucose, but appeared to have a predominant role in the regulation of postprandial glucose level in GIPR-/- mice compared with GLP-1R-/- mice [34,35]. Furthermore, in clinical trials, GIP and GLP-1 induced the similar early phase insulin responses to oral glucose challenge, but GIP failed to induce the late phase insulin response in diabetic patients [36].
BIOLOGICAL ACTIONS OF GLP-1 AND GIP IN β-CELLS
- Meal ingestion and autonomic nervous system
- GLP-1 and GIP release can be stimulated by mixed meals or individual nutrients including glucose and fatty acids [37]. GIP secretion showed species-specific differences, in which fat is the most potent stimulator in humans and carbohydrate is the most potent stimulator in rodents and pigs [11].
- Glucose entry into the L-cells via sodium glucose transporters (SGLT) causes an increase in ATP level, which leads to the closure of KATP channels and subsequent opening of L-type voltage-gated calcium channels [38], which results in GLP-1 release. Fatty acids bound to GPR120 increase the intracellular calcium level and activate AKT or MAPK signaling, which lead to GLP-1 release. However, the nutrient-stimulated GLP-1 release starts at a second phase (90-120 min) after nutrient ingestion [12]. In the early phase (15-30 min) after nutrient ingestion, GLP-1 secretion seems to be mainly stimulated by the autonomic nervous system, by neurotransmitters gastrin-releasing peptide (GRP) and acetylcholine (Ach), and GIP, because the majority of GLP-1 secreting L-cells are located in the distal small intestine [12]. After a meal ingestion, GIP released in the duodenum appears to activate the vagal afferents, which subsequently induce GLP-1 secretion through the vagal efferents (Ach) or enteric efferent neurons such as GRP. Binding of GRP or Ach to G protein-linked GPR or M1 receptors activates phospholipase C (PLC)/protein kinase C (PKC) or cAMP/PKA signaling and then leads to secretion of GLP-1. The importance of the vagus nerve in mediating the proximal-distal loop was elucidated from the evidence that GLP-1 secretion is enhanced when the fat is administered into the duodenum or when the GLP-1 secretion in response to the infusion of physiological concentration of GIP was completely abrogated by vagotomy [39].
- Leptin
- In leptin deficient ob/ob mice and high fat-induced leptin resistant mice, plasma GLP-1 levels were decreased. Binding of leptin to its receptor (OB-Rb) leads to phosphorylation of Janus kinase (JAK), which serves as a docking site for a signal transducer and activator of transcription (STAT) molecules. Once docked, STAT molecules are phosphorylated and dimerized prior to entering into the nucleus to mediate gene transcription. In human and rodent L-cells, leptin increases GLP-1 secretion together with STAT3 phosphorylation. However, the detailed mechanisms on downstream mediators are not known. Nonetheless, these findings suggest a possible mechanism by which circulating GLP-1 levels are reduced in obese individuals [40].
- Activator of Wnt signaling: insulin and metformin
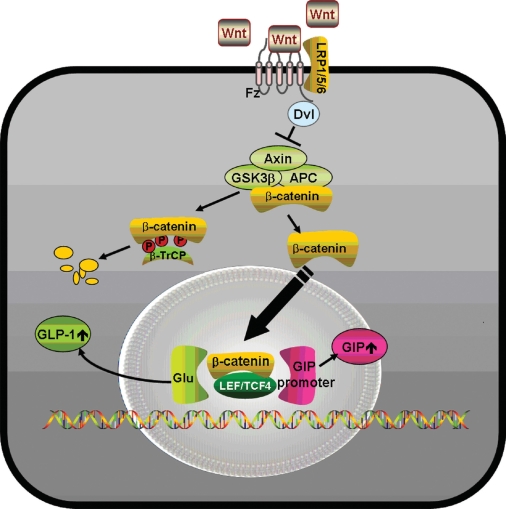
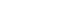
- Most, but not all studies have found that insulin secretion tends to decrease in carriers of the TCF4 (TCF7L2) risk genotypes, which implies potential mechanisms relating Wnt signaling to pancreatic development and β-cell function, as well as the regulation of the incretin hormones [41,42]. In carriers of TCF4 gene polymorphisms, GLP-1 administration did not induce insulin secretion [43] and Wnt3 or lithium, an inhibitor of glycogen synthase kinase (GSK) 3β, enhanced GIP production through the increase in the binding of β-catenin/TCF4 to the proximal GIP promoter [44]. In the presence of Wnt ligands, Wnts bind to Frizzled (Fz) receptors and low-density lipoprotein receptor-related protein (LRP) 5/6 coreceptors, whereby dishevelled (Dsh) is recruited to the membrane and GSK 3β is inhibited by the activation of Dsh by Fz. This could lead to the translocation of the β-catenin into the nucleus and could stimulate the binding of β-catenin with TCF to activate specific genes, such as GLP-1 and GIP (Fig. 1) [45].
- Insulin stimulates GLP-1 secretion via the cross-talk between insulin and Wnt signaling pathways. In an intestinal GLUTag L-cell line as well as in primary fetal rat intestinal cells, insulin stimulated the expression of proglucagon and enhanced GLP-1 content, which is attributable to the enhanced binding of β-catenin and TCF-4 to the TCF4 site in the G2 enhancer element [46] of the proglucagon gene promoter. Moreover, a high concentration of insulin can induce insulin resistance in L-cells and this insulin resistance can attenuate insulin-stimulated GLP-1 secretion as well as heterologous desensitization of the L-cell response to GIP. Taken together, these findings suggest a possible mechanism by which GLP-1 levels could be reduced in patients with type 2 diabetes [47] as well as in normal subjects in the lowest tertile of insulin sensitivity [12]. Recently, we found that metformin could increase the production and secretion of GLP-1 in intestinal L-cells both in vitro and in vivo through the activation of the Wnt signal transduction pathway (unpublished data).
- A variety of genes bind to the GIP gene promoter
- Human GIPR gene promoter contains cAMP-response elements and the binding sites for several transcription factors including Sp1, Sp3, activator protein (AP)-1 and AP-2. In addition, cis-acting negative regulatory sequences that control cell-specific GIPR gene expression have been identified in more distal 5'-flanking region [48]. Recently, chromatin immunoprecipitation assay was used to demonstrate that Pdx-1 binds to GIP promoter. Indeed, there was a remarkable reduction (97.8%) in the number of GIP-expressing cells in Pdx-/- mice [49].
FACTORS INVOLVED IN GLP-1 AND GIP SYNTHESIS AND SECRETION
- GLP-1 and GIP could exert beneficial effects in β-cells through structurally related G protein-coupled receptors, which utilize overlapping signal transduction pathways in islet β-cells. Although GLP-1 and GIP both stimulate glucose-dependent insulin secretion, insulin synthesis, and β-cell proliferation, and inhibit β-cell apoptosis, they exert different activities and actions on β-cells, too. Thus, understanding the relative importance and the different mechanisms of incretin hormones on β-cells will help us to design strategies to optimize β-cell function in patients with T2DM.
CONCLUSION
- 1. World Health Organization: Diabetes facts (online) cited 2010 Feb 8. Available from: http://www.who.int/mediacentre/fatsheets/fs312/en/index.htm.
- 2. International Diabetes Federation (IDF): IDF diabetes Atlas cited 2010 Feb 8. Available from: http://www.diabetesatlas.org/map.
- 3. Yoon KH, Lee JH, Kim JW, Cho JH, Choi YH, Ko SH, Zimmet P, Son HY. Epidemic obesity and type 2 diabetes in Asia. Lancet 2006;368:1681-1688. ArticlePubMed
- 4. U.K. Prospective Diabetes Study Group. U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. Diabetes 1995;44:1249-1258. ArticlePubMed
- 5. Gerich JE. Redefining the clinical management of type 2 diabetes: matching therapy to pathophysiology. Eur J Clin Invest 2002;32(Suppl 3):46-53. ArticlePubMedPDF
- 6. Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC, Palmer JP. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects: evidence for a hyperbolic function. Diabetes 1993;42:1663-1672. ArticlePubMed
- 7. Kim DJ, Lee MS, Kim KW, Lee MK. Insulin secretory dysfunction and insulin resistance in the pathogenesis of Korean type 2 diabetes mellitus. Metabolism 2001;50:590-593. ArticlePubMed
- 8. Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, Son HY, Kang SK, Kim HS, Lee IK, Bonner-Weir S. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003;88:2300-2308. PubMed
- 9. Shin CS, Lee HK, Koh CS, Kim YI, Shin YS, Yoo KY, Paik HY, Park YS, Yang BG. Risk factors for the development of NIDDM in Yonchon County, Korea. Diabetes Care 1997;20:1842-1846. ArticlePubMedPDF
- 10. Kuroe A, Fukushima M, Usami M, Ikeda M, Nakai Y, Taniguchi A, Matsuura T, Suzuki H, Kurose T, Yasuda K, Yamada Y, Seino Y. Impaired beta-cell function and insulin sensitivity in Japanese subjects with normal glucose tolerance. Diabetes Res Clin Pract 2003;59:71-77. PubMed
- 11. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007;132:2131-2157. ArticlePubMed
- 12. Rask E, Olsson T, Soderberg S, Johnson O, Seckl J, Holst JJ, Ahren B. Impaired incretin response after a mixed meal is associated with insulin resistance in nondiabetic men. Diabetes Care 2001;24:1640-1645. ArticlePubMedPDF
- 13. Lugari R, Dell'Anna C, Ugolotti D, Dei Cas A, Barilli AL, Zandomeneghi R, Marani B, Iotti M, Orlandini A, Gnudi A. Effect of nutrient ingestion on glucagon-like peptide 1 (7-36 amide) secretion in human type 1 and type 2 diabetes. Horm Metab Res 2000;32:424-428. ArticlePubMed
- 14. Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, Lyssenko V, Bouatia-Naji N, Dupuis J, Jackson AU, Kao WH, Li M, Glazer NL, Manning AK, Luan J, Stringham HM, Prokopenko I, Johnson T, Grarup N, Boesgaard TW, Lecoeur C, Shrader P, O'Connell J, Ingelsson E, Couper DJ, Rice K, Song K, Andreasen CH, Dina C, Kottgen A, Le Bacquer O, Pattou F, Taneera J, Steinthorsdottir V, Rybin D, Ardlie K, Sampson M, Qi L, van Hoek M, Weedon MN, Aulchenko YS, Voight BF, Grallert H, Balkau B, Bergman RN, Bielinski SJ, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Bottcher Y, Brunner E, Buchanan TA, Bumpstead SJ, Cavalcanti-Proenca C, Charpentier G, Chen YD, Chines PS, Collins FS, Cornelis M, G JC, Delplanque J, Doney A, Egan JM, Erdos MR, Firmann M, Forouhi NG, Fox CS, Goodarzi MO, Graessler J, Hingorani A, Isomaa B, Jorgensen T, Kivimaki M, Kovacs P, Krohn K, Kumari M, Lauritzen T, Levy-Marchal C, Mayor V, McAteer JB, Meyre D, Mitchell BD, Mohlke KL, Morken MA, Narisu N, Palmer CN, Pakyz R, Pascoe L, Payne F, Pearson D, Rathmann W, Sandbaek A, Sayer AA, Scott LJ, Sharp SJ, Sijbrands E, Singleton A, Siscovick DS, Smith NL, Sparso T, Swift AJ, Syddall H, Thorleifsson G, Tonjes A, Tuomi T, Tuomilehto J, Valle TT, Waeber G, Walley A, Waterworth DM, Zeggini E, Zhao JH, Illig T, Wichmann HE, Wilson JF, van Duijn C, Hu FB, Morris AD, Frayling TM, Hattersley AT, Thorsteinsdottir U, Stefansson K, Nilsson P, Syvanen AC, Shuldiner AR, Walker M, Bornstein SR, Schwarz P, Williams GH, Nathan DM, Kuusisto J, Laakso M, Cooper C, Marmot M, Ferrucci L, Mooser V, Stumvoll M, Loos RJ, Altshuler D, Psaty BM, Rotter JI, Boerwinkle E, Hansen T, Pedersen O, Florez JC, McCarthy MI, Boehnke M, Barroso I, Sladek R, Froguel P, Meigs JB, Groop L, Wareham NJ, Watanabe RM. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010;42:142-148. ArticlePubMedPMCPDF
- 15. Lee S, Yabe D, Nohtomi K, Takada M, Morita R, Seino Y, Hirano T. Intact glucagon-like peptide-1 levels are not decreased in Japanese patients with type 2 diabetes. Endocr J 2010;57:119-126. ArticlePubMed
- 16. Moore B. On the treatment of diabetus mellitus by acid extract of duodenal mucous membrane. Biochem J 1906;1:28-38. ArticlePubMedPMCPDF
- 17. McIntyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet 1964;2:20-21. Article
- 18. Andersen DK, Elahi D, Brown JC, Tobin JD, Andres R. Oral glucose augmentation of insulin secretion: interactions of gastric inhibitory polypeptide with ambient glucose and insulin levels. J Clin Invest 1978;62:152-161. ArticlePubMedPMC
- 19. Ebert R, Unger H, Creutzfeldt W. Preservation of incretin activity after removal of gastric inhibitory polypeptide (GIP) from rat gut extracts by immunoadsorption. Diabetologia 1983;24:449-454. ArticlePubMedPDF
- 20. Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem 1986;261:11880-11889. ArticlePubMed
- 21. Orskov C, Holst JJ, Poulsen SS, Kirkegaard P. Pancreatic and intestinal processing of proglucagon in man. Diabetologia 1987;30:874-881. ArticlePubMedPDF
- 22. Furuta M, Yano H, Zhou A, Rouille Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci U S A 1997;94:6646-6651. ArticlePubMedPMC
- 23. Mortensen K, Christensen LL, Holst JJ, Orskov C. GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept 2003;114:189-196. ArticlePubMed
- 24. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995;136:3585-3596. ArticlePubMed
- 25. Montrose-Rafizadeh C, Avdonin P, Garant MJ, Rodgers BD, Kole S, Yang H, Levine MA, Schwindinger W, Bernier M. Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology 1999;140:1132-1140. ArticlePubMed
- 26. Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 1993;133:2861-2870. ArticlePubMed
- 27. Holz GG. Epac: a new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes 2004;53:5-13. PubMedPMC
- 28. Wang X, Cahill CM, Pineyro MA, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 regulates the beta cell transcription factor, PDX-1, in insulinoma cells. Endocrinology 1999;140:4904-4907. ArticlePubMed
- 29. Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. Beta-cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes 2005;54:482-491. PubMed
- 30. Hui H, Wright C, Perfetti R. Glucagon-like peptide 1 induces differentiation of islet duodenal homeobox-1-positive pancreatic ductal cells into insulin-secreting cells. Diabetes 2001;50:785-796. ArticlePubMedPDF
- 31. Buteau J, Spatz ML, Accili D. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass. Diabetes 2006;55:1190-1196. PubMed
- 32. Yusta B, Baggio LL, Estall JL, Koehler JA, Holland DP, Li H, Pipeleers D, Ling Z, Drucker DJ. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 2006;4:391-406. PubMed
- 33. Kim SJ, Choi WS, Han JS, Warnock G, Fedida D, McIntosh CH. A novel mechanism for the suppression of a voltage-gated potassium channel by glucose-dependent insulinotropic polypeptide: protein kinase A-dependent endocytosis. J Biol Chem 2005;280:28692-28700. PubMed
- 34. Miyawaki K, Yamada Y, Yano H, Niwa H, Ban N, Ihara Y, Kubota A, Fujimoto S, Kajikawa M, Kuroe A, Tsuda K, Hashimoto H, Yamashita T, Jomori T, Tashiro F, Miyazaki J, Seino Y. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci U S A 1999;96:14843-14847. ArticlePubMedPMC
- 35. Pamir N, Lynn FC, Buchan AM, Ehses J, Hinke SA, Pospisilik JA, Miyawaki K, Yamada Y, Seino Y, McIntosh CH, Pederson RA. Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am J Physiol Endocrinol Metab 2003;284:E931-E939. ArticlePubMed
- 36. Vilsboll T. On the role of the incretin hormones GIP and GLP-1 in the pathogenesis of type 2 diabetes mellitus. Dan Med Bull 2004;51:364-370. PubMed
- 37. Brubaker PL. The glucagon-like peptides: pleiotropic regulators of nutrient homeostasis. Ann N Y Acad Sci 2006;1070:10-26. PubMed
- 38. Lim GE, Brubaker PL. Glucagon-like peptide 1 secretion by the L-cell. Diabetes 2006;55(Suppl 2):S70-S77.ArticlePDF
- 39. Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology 1999;140:1687-1694. ArticlePubMed
- 40. Anini Y, Brubaker PL. Role of leptin in the regulation of glucagon-like peptide-1 secretion. Diabetes 2003;52:252-259. ArticlePubMedPDF
- 41. Elbein SC, Chu WS, Das SK, Yao-Borengasser A, Hasstedt SJ, Wang H, Rasouli N, Kern PA. Transcription factor 7-like 2 polymorphisms and type 2 diabetes, glucose homeostasis traits and gene expression in US participants of European and African descent. Diabetologia 2007;50:1621-1630. ArticlePubMedPDF
- 42. Wang J, Kuusisto J, Vanttinen M, Kuulasmaa T, Lindstrom J, Tuomilehto J, Uusitupa M, Laakso M. Variants of transcription factor 7-like 2 (TCF7L2) gene predict conversion to type 2 diabetes in the Finnish Diabetes Prevention Study and are associated with impaired glucose regulation and impaired insulin secretion. Diabetologia 2007;50:1192-1200. ArticlePubMedPDF
- 43. Schafer SA, Tschritter O, Machicao F, Thamer C, Stefan N, Gallwitz B, Holst JJ, Dekker JM, t Hart LM, Nijpels G, van Haeften TW, Haring HU, Fritsche A. Impaired glucagon-like peptide-1-induced insulin secretion in carriers of transcription factor 7-like 2 (TCF7L2) gene polymorphisms. Diabetologia 2007;50:2443-2450. ArticlePubMedPMCPDF
- 44. Garcia-Martinez JM, Chocarro-Calvo A, Moya CM, Garcia-Jimenez C. WNT/beta-catenin increases the production of incretins by entero-endocrine cells. Diabetologia 2009;52:1913-1924. ArticlePubMedPDF
- 45. Gustafson B, Smith U. Wnt signaling is both an inducer and effector of glucagon-like peptide-1. Diabetologia 2008;51:1768-1770. ArticlePubMedPDF
- 46. Yi F, Sun J, Lim GE, Fantus IG, Brubaker PL, Jin T. Cross talk between the insulin and Wnt signaling pathways: evidence from intestinal endocrine L cells. Endocrinology 2008;149:2341-2351. ArticlePubMed
- 47. Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, Holst JJ. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001;86:3717-3723. ArticlePubMed
- 48. Lynn FC, Pamir N, Ng EH, McIntosh CH, Kieffer TJ, Pederson RA. Defective glucose-dependent insulinotropic polypeptide receptor expression in diabetic fatty Zucker rats. Diabetes 2001;50:1004-1011. ArticlePubMedPDF
- 49. Jepeal LI, Fujitani Y, Boylan MO, Wilson CN, Wright CV, Wolfe MM. Cell-specific expression of glucose-dependent-insulinotropic polypeptide is regulated by the transcription factor PDX-1. Endocrinology 2005;146:383-391. ArticlePubMedPDF
REFERENCES
Fig. 1Proposed model for action of Wnt signaling on GLP-1 or GIP production. Wnt ligands bind to Frizzled (Fz) receptors and low-density lipoprotein receptor-related protein (LRP) 5/6 coreceptors, whereby dishevelled (Dsh) is recruited to the membrane and GSK3β is inhibited by the activation of Dsh by Fz. This leads to the translocation of β-catenin into the nucleus, where it binds with TCF to activate specific target genes such as GLP-1 and GIP in intestinal L-cells or K-cells. GLP-1, glucagon-like peptide-1; GIP, glucose-dependent insulinotropic polypeptide.
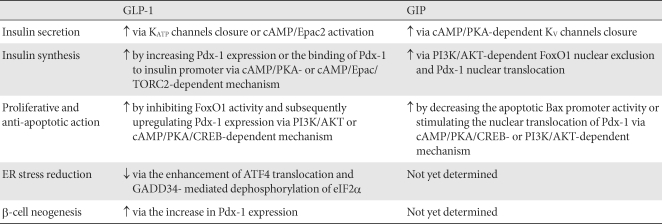
Figure & Data
References
Citations
Citations to this article as recorded by 
- A phase I study comparing the pharmacokinetics of the biosimilar (RD12014) with liraglutide (Victoza) in healthy Chinese male subjects
Ruirui Zhou, Linfeng Guo, Xianglei Gao, Yijun Wang, Wenjing Xu, Yang Zou, Wenjia Li, Yulei Zhuang, Gangyi Liu, Yanmei Liu
Clinical and Translational Science.2022; 15(10): 2458. CrossRef - Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases
Mustafa Karabicici, Yagmur Azbazdar, Evin Iscan, Gunes Ozhan
Membranes.2021; 11(11): 844. CrossRef - Effects of newly introduced antidiabetic drugs on autophagy
Milad Ashrafizadeh, Habib Yaribeygi, Stephen L. Atkin, Amirhossein Sahebkar
Diabetes & Metabolic Syndrome: Clinical Research & Reviews.2019; 13(4): 2445. CrossRef - Genistein enhances the secretion of glucagon-like peptide-1 (GLP-1) via downregulation of inflammatory responses
Kanwal Rehman, Mehwish Bagh Ali, Muhammad Sajid Hamid Akash
Biomedicine & Pharmacotherapy.2019; 112: 108670. CrossRef - Liraglutide vs Exenatide: Patient Adherence, Medication Persistence and Economic Evaluation in the Treatment of Type 2 Diabetes Mellitus
Fiorenzo Santoleri, Paola Sorice, Ruggero Lasala, Alberto Costantini
Pharmacology & Pharmacy.2014; 05(04): 332. CrossRef - Metformin enhances glucagon-like peptide 1 via cooperation between insulin and Wnt signaling
Mi-Hyun Kim, Jae-Hwan Jee, Sunyoung Park, Myung-Shik Lee, Kwang-Won Kim, Moon-Kyu Lee
Journal of Endocrinology.2014; 220(2): 117. CrossRef - Predicting the DPP-IV Inhibitory ActivitypIC50Based on Their Physicochemical Properties
Tianhong Gu, Xiaoyan Yang, Minjie Li, Milin Wu, Qiang Su, Wencong Lu, Yuhui Zhang
BioMed Research International.2013; 2013: 1. CrossRef - Exendin-4 Protects Against Sulfonylurea-Induced β-Cell Apoptosis
Ju-Young Kim, Dong-Mee Lim, Hyung-Seo Park, Chan-Il Moon, Kyung-Jin Choi, Seong-Kyu Lee, Haing-Woon Baik, Keun-Young Park, Byung-Joon Kim
Journal of Pharmacological Sciences.2012; 118(1): 65. CrossRef - Retrospective Analysis on the Efficacy, Safety and Treatment Failure Group of Sitagliptin for Mean 10-Month Duration
Won Jun Kim, Cheol-Young Park, Eun Haeng Jeong, Jeong Youn Seo, Ji Soo Seol, Se Eun Park, Eun Jung Rhee, Won Young Lee, Ki Won Oh, Sung Woo Park, Sun Woo Kim
Diabetes & Metabolism Journal.2011; 35(3): 290. CrossRef
 PubReader
PubReader Cite
Cite