- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 46(1); 2022 > Article
-
ReviewIslet Studies and Transplantation Regulation of Pancreatic β-Cell Mass by Gene-Environment Interaction
-
Shun-ichiro Asahara1
 , Hiroyuki Inoue2, Yoshiaki Kido1,2
, Hiroyuki Inoue2, Yoshiaki Kido1,2 -
Diabetes & Metabolism Journal 2022;46(1):38-48.
DOI: https://doi.org/10.4093/dmj.2021.0045
Published online: January 27, 2022
1Division of Diabetes and Endocrinology, Department of Internal Medicine, Kobe University Graduate School of Medicine, Kobe, Japan
2Division of Medical Chemistry, Department of Metabolism and Diseases, Kobe University Graduate School of Health Sciences, Kobe, Japan
-
Corresponding author: Yoshiaki Kido Department of Metabolism and Diseases, Kobe University Graduate School of Health Sciences, 7-10-2 Tomogaoka, Suma-ku, Kobe 654-0142, Japan E-mail: kido@med.kobe-u.ac.jp
• Received: March 15, 2021 • Accepted: May 28, 2021
Copyright © 2022 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- Graphical abstract
- INTRODUCTION
- PHENOTYPIC VARIATIONS AMONG MOUSE STRAINS AND HUMAN ETHNIC GROUPS
- DEVELOPMENTAL ORIGINS OF HEALTH AND DISEASE
- EPIGENETICS
- TYPE 1 DIABETES MELLITUS AND GENE-ENVIRONMENT INTERACTIONS
- INTERACTIONS BETWEEN DIABETES SUSCEPTIBILITY GENES AND HFD FEEDING
- CONCLUSIONS
- NOTES
- REFERENCES
ABSTRACT
- The main pathogenic mechanism of diabetes consists of an increase in insulin resistance and a decrease in insulin secretion from pancreatic β-cells. The number of diabetic patients has been increasing dramatically worldwide, especially in Asian people whose capacity for insulin secretion is inherently lower than that of other ethnic populations. Causally, changes of environmental factors in addition to intrinsic genetic factors have been considered to have an influence on the increased prevalence of diabetes. Particular focus has been placed on “gene-environment interactions” in the development of a reduced pancreatic β-cell mass, as well as type 1 and type 2 diabetes mellitus. Changes in the intrauterine environment, such as intrauterine growth restriction, contribute to alterations of gene expression in pancreatic β-cells, ultimately resulting in the development of pancreatic β-cell failure and diabetes. As a molecular mechanism underlying the effect of the intrauterine environment, epigenetic modifications have been widely investigated. The association of diabetes susceptibility genes or dietary habits with gene-environment interactions has been reported. In this review, we provide an overview of the role of gene-environment interactions in pancreatic β-cell failure as revealed by previous reports and data from experiments.
- The incidence of diabetes has been increasing rapidly for several decades worldwide. There were more than 400 million patients with diabetes in 2019, and this number is estimated to reach 700 million by 2045 [1]. In the Western Pacific region, including East Asia, there was an especially large increase in the number of diabetic patients after World War II because of dietary changes and a decrease in physical activity. East Asian individuals have lower insulin secretory capacity than Caucasians; thus, East Asians are considered to have a tendency to develop pancreatic β-cell failure owing to genetic factors [2-4].
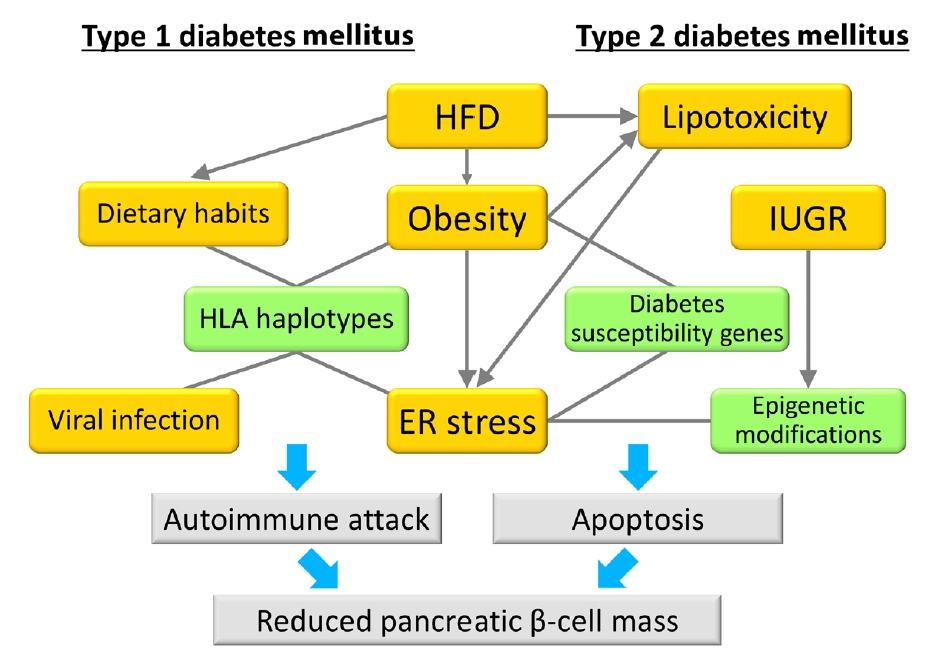
- Previous work has shown the importance of insulin signaling in pancreatic β-cells using β-cell-specific transgenic mice. In 2006, Hashimoto et al. [5] clarified that a deficiency of 3-phosphoinositide dependent protein kinase 1 (Pdk1), an insulin signaling-related molecule, resulted in severe pancreatic β-cell failure, leading to hyperglycemia. In addition, it has been reported that mice deficient for tuberous sclerosis complex 2, a downstream molecule of Pdk1, exclusively in pancreatic β-cells develop hyperinsulinemia as a result of increased β-cell mass at younger ages, but show downregulation of insulin signaling induced by autophagic impairment and negative feedback, resulting in pancreatic β-cell failure at older ages [6-8]. To date, many investigators have used genetically modified mice as the main tool for the study of β-cell failure. These studies have revealed the pathogenesis of diabetes and the important role of various molecules in the onset of pancreatic β-cell failure. However, diabetes is a multifactorial disease. There are only a few monogenic forms of diabetes, such as maturity onset diabetes of the young, and it is considered that pancreatic β-cell failure as well as diabetes generally occur through a combination of genetic and environmental factors. The rapid increase in the number of diabetic patients in East Asia must be the result of genetic factors inherent in Asian people and environmental factors that have changed recently. Furthermore, for the combination of genetic and environmental factors, synergistic effects are considered to be more important than additive effects for the onset of diabetes and pancreatic β-cell failure. Thus, environmental factors increase the influence of some genetic factors synergistically, resulting in the development of diabetes and pancreatic β-cell failure (Fig. 1). Understanding gene-environment interactions is expected to lead to the personalization of diet and exercise therapy. In this review, we describe gene-environment interactions according to the findings of previous studies.
INTRODUCTION
- First, we discuss the effects of diet in different mouse strains as a simple example of gene-environment interactions. Currently, C57BL/6J is the most widely used mouse strain in animal experiments because this strain readily shows phenomena such as obesity with a high-fat diet (HFD) [9,10]. C57BL/6J mice are lean with a normal chow diet, but they develop hyperglycemia, hyperinsulinemia, and hyperleptinemia with a HFD [11-13]. On the other hand, strains such as 129/Sv or A/J are resistant to obesity and hyperglycemia under a HFD [14]. When C57BL/6J and 129/Sv mice are fed an HFD, 129/Sv mice show an increase in thermogenesis and maintain insulin sensitivity. When the genetic backgrounds of both strains were compared by a genome-wide scan, there was a significant difference in a locus associated with insulin resistance located on chromosome 14 [15]. Namely, the genetic differences between these strains are considered to produce their divergent phenotypic responses to a HFD.
- Such phenomena are also observed in humans, with differences in body mass index, energy consumption, insulin resistance, and insulin secretion between East Asian and Caucasian populations [16-20]. Moreover, it has been recently reported that the regulation of pancreatic β-cell mass varies among racial groups [21-24]. Saisho et al. [21] found that pancreatic β-cell mass was increased in an obese group of healthy Caucasians when compared with a lean group, which was considered to be due to a compensatory mechanism for insulin resistance. Conversely, when pancreatic β-cell mass was quantified in healthy Japanese subjects and compared between obese and lean groups, there was no significant difference in pancreatic β-cell mass between the groups, but there was a slight decrease in the obese group compared with the lean group [22]. These results indicate that the compensatory mechanisms for pancreatic β-cell mass under conditions of insulin resistance, such as obesity, are different between Caucasian and Japanese individuals. It is considered that the phenotype may differ according to genetic factors, even if the dietary habits are the same.
PHENOTYPIC VARIATIONS AMONG MOUSE STRAINS AND HUMAN ETHNIC GROUPS
- Environmental factors can affect genetic factors in adulthood, but the most important gene-environment interactions occur during the fetal period. In 1986, Barker and Osmond [25] proposed a theory that malnutrition in early life, including fetal life, causes susceptibility to ischemic heart disease and diabetes in adulthood (Barker’s hypothesis). This hypothesis, which is also referred to as the Developmental Origins of Health and Disease theory, suggests that the embryonic or neonatal environment significantly contributes to the onset of future metabolic diseases [26-30]. A 50-year epidemiological survey reported that children of mothers who experienced the “Dutch famine” at the end of World War II have a high incidence of hypertension, obesity, or glucose intolerance [31,32]. Furthermore, the mechanism underlying this phenomenon has been elucidated using animal models. Yura et al. [33] generated intrauterine growth restriction (IUGR) mice with uterine artery ligation as a model of undernutrition. They showed that IUGR mice have significantly lower birth weight than control mice, but there is no significant difference in body weight in adulthood between both groups because IUGR mice show “catchup” growth after weening. In addition, when both groups were fed a HFD, IUGR mice exhibit significantly more severe obesity and glucose intolerance [33]. This is considered to be due to a “neonatal leptin surge,” that is, an increase in serum leptin levels during the neonatal period, which is considered to lead to the onset of obesity by inducing leptin resistance.
- Other reports have indicated that catch-up growth is associated with the development of obesity in later life in humans and mice [34-38]. Catch-up growth is considered to contribute significantly to the development of metabolic syndrome. It has previously been reported that catch-up growth occurs in pancreatic β-cell mass as well as the whole body in the adult period and insulin signaling has a key mechanistic role in this process [39,40]. In addition, the offspring of female rats lacking vitamin B12 have been reported to be susceptible to insulin resistance in adulthood, and the offspring of pregnant rats fed a low-protein diet also develop insulin resistance through the reduction of protein kinase C zeta (PKCζ) expression in skeletal muscle [41-43].
- Recently, an increase in fat intake has become a severe problem in developed countries. The offspring of HFD-fed female monkeys exhibit increased expression of gluconeogenic enzymes and transcription factors in hepatic tissues, resulting in the development of nonalcoholic fatty liver disease [44]. Interestingly, the female offspring of male HFD-fed rats exhibit pancreatic β-cell failure and hyperglycemia [45]. Compared with control mice, the female offspring of HFD-fed fathers show a change in the expression of 642 genes in microarray analysis using pancreatic islets. These changes were considered to induce pancreatic β-cell failure.
- As described above, many reports have described the effects of fetal and neonatal environments on genetic factors, leading to type 2 diabetes mellitus (T2DM) and obesity. The most wellknown molecular mechanism underlying this process is epigenetics, and we discuss the association between T2DM and epigenetics in the next section.
DEVELOPMENTAL ORIGINS OF HEALTH AND DISEASE
- Epigenetics is regarded as an important molecular mechanism in not only metabolic diseases but also various other diseases. Epigenetics is a system that changes gene expression via DNA or histone modifications without mutation of the nucleotide sequence [46-48]. As it is considered that environmental changes can affect gene expression, we should not forget this mechanism when we consider gene-environment interactions. In particular, changes in the intrauterine environment can have an impact on epigenetic modifications, and fetal nutritional conditions memorized in embryonic tissue have been found to lead to the development of various diseases in the future (metabolic memory) [49-51]. Park et al. [52] generated an IUGR rat model by ligation of the uterine artery, and these rats developed pancreatic β-cell failure. When they analyzed epigenetic modifications in isolated islets of IUGR rats, they found that the expression of pancreatic and duodenal homeobox 1 (Pdx1), a transcription factor, was decreased through an increase in DNA methylation and a change of histone modification at the Pdx1 promoter. The offspring of low-protein diet-fed female mice developed T2DM in adulthood, and there was a reduction of hepatocyte nuclear factor 4 alpha (HNF4α) expression in the pancreatic islets of those mice. This phenomenon was shown to be due to the inhibition of HNF4α promoter-enhancer interactions [53]. In addition, in stem cells isolated from cord blood of neonates with IUGR, changes of DNA methylation in multiple genes including HNF4α were found [54]. IUGR was predicted to influence the expression of various transcription factors, regardless of species or tissue.
- The effects of IUGR have also been investigated in other insulin target organs [55,56], for instance, histone modifications at the glucose transporter type 4 (Glut4) promoter contribute to a reduction in Glut4 expression through the suppression of transcription in the skeletal muscle of IUGR rats [57]. Furthermore, in hepatocytes, Ehara et al. [58] clarified that the expression of glycerol-3-phosphate acyltransferase 1 was regulated by DNA methylation of its promoter. Interestingly, the expression of lipogenic genes is affected in the male offspring (F1) of IUGR mice, and even in hepatocytes of F2 mice. The expression of liver X receptor-alpha (Lxra) and DNA methylation at the Lxra promoter in hepatocytes of F2 mice has been reported to be altered significantly [59]. The fact that the effect of IUGR was passed on not only to children but also to grandchildren was a huge surprise. Possibly, the effects of the Dutch famine after World War II may still remain today.
EPIGENETICS
- Type 1 diabetes mellitus (T1DM) is induced through the destruction of pancreatic β-cells by the autoimmune response [60,61], and it is likely to occur during childhood [62]. However, a number of individuals develop T1DM in adulthood [63]. Two large epidemiological studies, Diabetes Mondiale (DIAMOND) and European Diabetes (EURODIAB), reported that the incidence of T1DM was increasing worldwide [64,65]. The reasons given for this included changes in environmental factors. Therefore, in this section, we describe the relationship between gene-environment interactions and T1DM.
- Specific human leukocyte antigen (HLA) alleles are genetic risk factors for the onset of T1DM [66] and influence its incidence and age of onset [67,68]. The highest risk HLA haplotypes are DRB1*03:01-DQA1*05:01-DQB1*02:01 (known as DR3) and DRB1*04-DQA1*03:01-DQB1*03:02 (known as DR4). Individuals heterozygous for DR3/4 have an odds ratio (OR) of 16.59 for T1DM, while DR3/3 and DR4/4 homozygotes have ORs of 6.32 and 5.68, respectively [66]. These ORs are considerably higher than those of T2DM susceptibility genes. However, it is difficult to explain how the number of T1DM patients is increased by genetic factors alone. In recent years, the number of obese people has increased gradually worldwide [69-71]. In addition, the prevalence of overweight and obesity has risen in children with T1DM [72,73]. The increase in the number of obese subjects may be associated with the increase in the number of patients with T1DM. Interestingly, in Finland and Norway, which are two high-incidence T1DM countries, a rate of increase of T1DM has plateaued in recent years [74], indicating that this phenomenon is due to a plateau in the growth of obesity in both countries [75]. The ratio of obese subjects in a T1DM group was shown to be nearly equivalent to that in the general population or underwent a rapid increase [76]. In recent reports, the risk of developing T1DM was 63% higher in obese children than in lean children [77], and obesity increased susceptibility to insulin resistance, resulting in stress on β-cells and rendering them more vulnerable to an autoimmune attack [78].
- Viral infection has been focused on as an important environmental factor of T1DM. It has been previously reported that enteroviruses, coxsackievirus B, cytomegalovirus, rotaviruses, and encephalomyocarditis virus contribute to the pathogenesis of T1DM [79,80]. In addition, dietary habits were found to be associated with the onset of T1DM as well as T2DM. Protein from cow milk and gluten are associated with an increase or decrease in the risk of developing T1DM [81-84].
- The mechanisms associated with the destruction of pancreatic β-cells by environmental factors have not been elucidated fully [85]. One possibility is that endoplasmic reticulum (ER) stress contributes to the destruction of β-cells [86]. Inflammatory cytokines released by insulitis disrupt ER homeostasis, resulting in the induction of the unfolded protein response. During this adaptive phase, activation of the inflammatory response is predicted to play an important role in the pathogenesis of T1DM [87]. Furthermore, the fact that ER stress induces translational errors suggests that aberrant insulin produced by ER stress may become a target of the autoimmune response as an antigenic peptide [88].
TYPE 1 DIABETES MELLITUS AND GENE-ENVIRONMENT INTERACTIONS
- Finally, we discuss recent findings about gene-environment interactions in T2DM. In 2008, the potassium voltage-gated channel subfamily Q member 1 (KCNQ1) gene was identified as a T2DM susceptibility gene through large-scale single nucleotide polymorphism analysis of Japanese T2DM patients [89,90]. Subsequent reports from many facilities confirmed that there is an association between KCNQ1 mutations and reduced insulin secretion [91-94], but the mechanism for the onset of diabetes remains unclear. It was previously reported that decreased expression of the non-coding RNA Kcnq1ot1, which is expressed from within the Kcnq1 genetic region, increases the expression of cyclin-dependent kinase inhibitor 1C (Cdkn1c) through epigenetic modifications, resulting in reduced pancreatic β-cell mass [95]. Kcnq1 is an imprinted gene, and Kcnq1ot1 expression is decreased in pancreatic β-cells of mice only when a Kcnq1 genetic mutation is paternally inherited. As a result, pancreatic β-cell mass is decreased by enhancing Cdkn1c expression. These results may partially indicate that Kcnq1 mutation is an underlying pathogenic mechanism of T2DM. However, the OR of developing T2DM was only 1.4, suggesting that T2DM is not induced by genetic mutation alone. Therefore, focus shifted to the fact that there is a CCAAT motif in the Cdkn1c promoter region. As the CCAAT motif is a binding site for members of the CCAAT enhancer-binding protein (C/EBP) transcription factor family, it was hypothesized that binding of C/EBP family members to this motif may further enhance Cdkn1c expression. C/EBPβ expression was found to be enhanced in the islets of db/db mice, a T2DM mouse model, and reported that the accumulation of C/EBPβ increased vulnerability to ER stress and resulted in reduced β-cell mass [96,97]. In addition, recent studies revealed that C/EBPβ also accumulates in the pancreatic islets of HFD-fed mice, which may cause pancreatic β-cell failure through enhanced Cdkn1c expression and the induction of ER stress, and is considered to contribute to the development of T2DM.
- Therefore, the influence of C/EBPβ accumulation in pancreatic β-cells was investigated when Kcnq1ot1 expression is decreased. A significant increase in fed blood glucose levels and a reduction in serum insulin levels were observed in mice with decreased Kcnq1ot1 expression and C/EBPβ overexpression exclusively in pancreatic β-cells, compared to control groups, which was accompanied by a significant decrease in pancreatic β-cell mass. This is presumed to be due to increased binding of C/EBPβ to the Cdkn1c promoter as a result of epigenetic modifications such as loosening chromatin structure under reduced Kcnq1ot1 expression. These results revealed that the synergistic effect of genetic factors (Kcnq1 mutation) and environmental factors (C/EBPβ accumulation) could induce markedly reduced pancreatic β-cell mass.
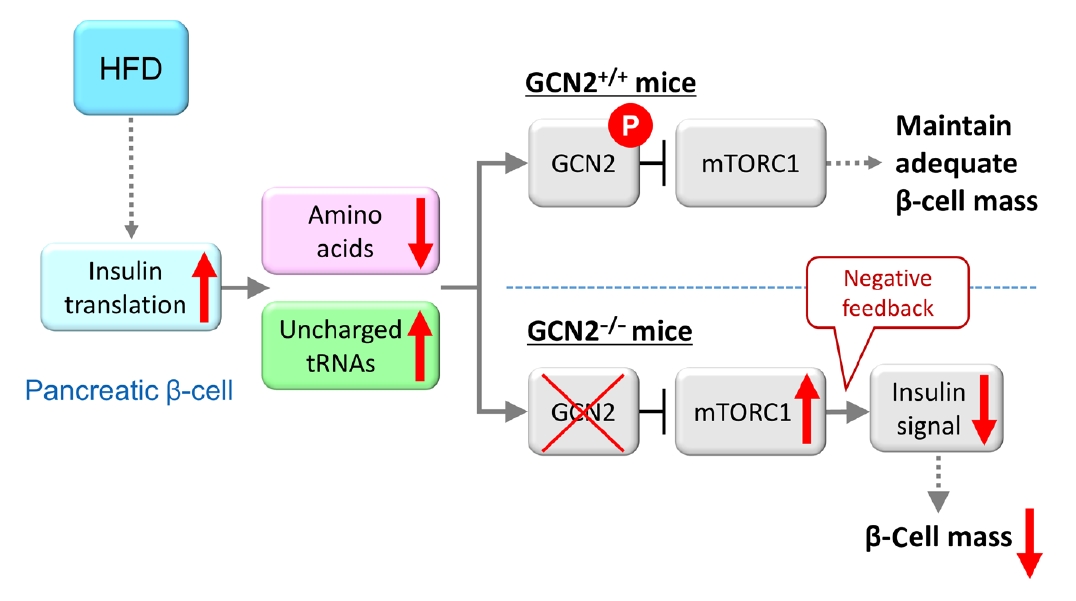
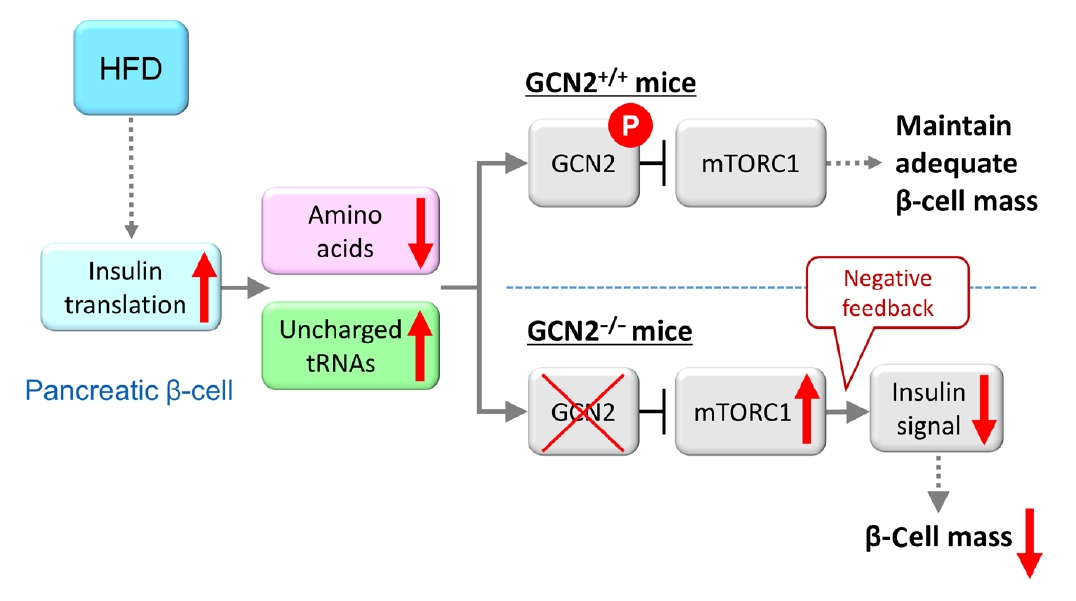
- Furthermore, the role of eukaryotic translation initiation factor 2α kinase 4 (EIF2AK4) was investigated. EIF2AK4 encodes general control nonderepressible 2 (GCN2), which, in yeast and mammals, is activated as a result of amino acid deficiency and was identified as a T2DM susceptibility gene, in addition to KCNQ1, through large-scale single nucleotide polymorphism analysis of Japanese T2DM patients [98-101]. It was found that insulin secretion was reduced in carriers of the EIF2AK4 risk allele compared with individuals without the risk allele [102]. In addition, HFD-fed GCN2 knockout mice exhibit reduced pancreatic β-cell mass and decreased insulin secretion, while the phenotype of GCN2 knockout mice fed a normal chow diet is comparable to that of control mice [102]. This phenomenon was attributed to the constitutive enhancement of mechanistic target of rapamycin complex 1 (mTORC1) activity in pancreatic β-cells of GCN2 knockout mice. In fact, chronic enhancement of mTORC1 signaling was shown to result in reduced pancreatic β-cell mass [6-8], and this mechanism was considered to contribute to the development of reduced β-cell mass in HFD-fed GCN2 knockout mice. GCN2 is known to be activated under cellular amino acid deficiency [99], and it was found that amino acid concentration was decreased in the pancreatic β-cells of HFD-fed mice [102]. The HFD was shown to increase insulin demand in the whole body, and a large quantity of amino acids was consumed due to insulin production in pancreatic β-cells [103], resulting in reduced amino acid concentrations in these cells [102]. In pancreatic β-cells of HFD-fed GCN2 knockout mice, GCN2 cannot be activated in spite of amino acid deficiency. HFD-fed GCN2 knockout mice were considered to show hyperglycemia accompanied by reduced pancreatic β-cell mass through the constitutive enhancement of mTORC1 due to GCN2 inactivation (Fig. 2). The effect of GCN2 alone is not sufficient to induce pancreatic β-cell failure; however, environmental factors such as a HFD interact with GCN2 synergistically to cause pancreatic β-cell failure. As far as previous studies have shown, pancreatic islets were the only tissue in which cellular amino acid levels were decreased [102]. This result indicates that the response of pancreatic β-cells to HFD feeding is unique, and that gene-environment interactions may have an especially strong influence on the viability of pancreatic β-cells, as many T2DM susceptibility genes are associated with insulin secretion. A recent report showed that pancreatic β-cell death caused by gene-environment interactions could be examined using human pluripotent stem cells [104]. This method may help to clarify the associations between diabetes susceptibility genes and gene-environment interactions.
INTERACTIONS BETWEEN DIABETES SUSCEPTIBILITY GENES AND HFD FEEDING
- The explosive increase in the number of diabetic patients worldwide in recent years is mainly due to changes in the environment, given that there have been no significant or consistent changes in genetic factors. However, changes in environmental factors are likely to affect genetic factors. In this review, we documented the evidence for the relationship between gene-environment interactions and IUGR or diabetes susceptibility genes. However, gene-environment interactions are believed to be universally applicable to the contemporary pathology of diabetes, especially reduced β-cell mass. Therefore, further detailed analysis of these gene-environment interactions may suggest the key for personalized medicine. We look forward to further progress in this field in the future.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
None
NOTES
-
Acknowledgements
- None
Fig. 1.Model of the associations between gene-environment interactions and pancreatic β-cell mass. In type 1 diabetes mellitus, an autoimmune attack is induced by the combination of genetic factors (human leukocyte antigen [HLA] haplotypes) and environmental factors (obesity, endoplasmic reticulum [ER] stress, dietary habits, and viral infection), resulting in a decrease in pancreatic β-cell mass. In type 2 diabetes mellitus, pancreatic β-cell mass is mainly regulated by the proliferation and apoptosis of pancreatic β-cells. Epigenetic modifications caused by intrauterine growth restriction (IUGR) are an important mechanism as a genetic factor. Environmental factors such as lipotoxicity, obesity, and ER stress caused by high-fat diet (HFD) feeding induce pancreatic β-cell failure in combination with genetic factors in a synergistic manner. Green boxes show genetic factors (nonmodifiable factors) and orange boxes show environmental factors (modifiable factors).
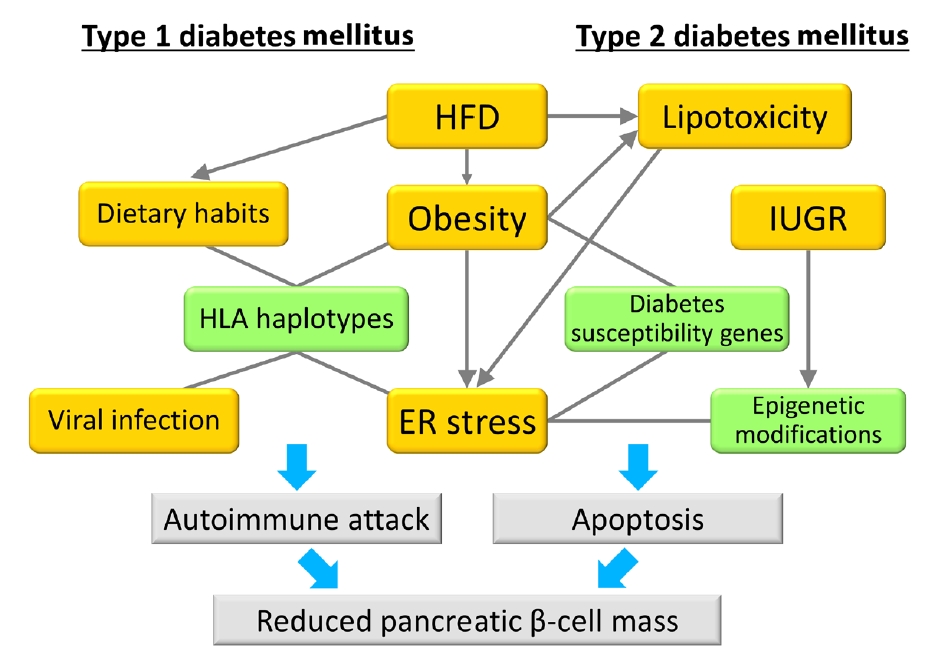
Fig. 2.Model for general control nonderepressible 2 (GCN2)-dependent regulation of pancreatic β-cell mass during high-fat diet (HFD) feeding. HFD feeding increases insulin demand, resulting in reduced amino acid concentrations due to their consumption in pancreatic β-cells. HFD-fed GCN2 knockout mice develop hyperglycemia accompanied by reduced pancreatic β-cell mass through the constitutive enhancement of mechanistic target of rapamycin complex 1 (mTORC1) due to GCN2 inactivation. GCN2, a genetic factor, and environmental factors, such as a HFD, cause pancreatic β-cell failure in a synergistic manner. “P” indicates the phosphorylation of GCN2. Modified from Kanno et al. [102], with permission from American Society for Clinical Investigation.
- 1. International Diabetes Federation. IDF Diabetes Atlas. 9th ed. Brussels: International Diabetes Federation; 2019.
- 2. Yabe D, Seino Y. Type 2 diabetes via β-cell dysfunction in East Asian people. Lancet Diabetes Endocrinol 2016;4:2-3.ArticlePubMed
- 3. Yabe D, Seino Y, Fukushima M, Seino S. β Cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes in East Asians. Curr Diab Rep 2015;15:602.ArticlePubMedPDF
- 4. Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in East Asians. Nat Genet 2011;44:67-72.PubMedPMC
- 5. Hashimoto N, Kido Y, Uchida T, Asahara S, Shigeyama Y, Matsuda T, et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat Genet 2006;38:589-93.ArticlePubMedPDF
- 6. Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol 2008;28:2971-9.PubMedPMC
- 7. Koyanagi M, Asahara S, Matsuda T, Hashimoto N, Shigeyama Y, Shibutani Y, et al. Ablation of TSC2 enhances insulin secretion by increasing the number of mitochondria through activation of mTORC1. PLoS One 2011;6:e23238.ArticlePubMedPMC
- 8. Bartolome A, Kimura-Koyanagi M, Asahara S, Guillen C, Inoue H, Teruyama K, et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014;63:2996-3008.ArticlePubMedPDF
- 9. Jo J, Gavrilova O, Pack S, Jou W, Mullen S, Sumner AE, et al. Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comput Biol 2009;5:e1000324.ArticlePubMedPMC
- 10. Kim DH, Gutierrez-Aguilar R, Kim HJ, Woods SC, Seeley RJ. Increased adipose tissue hypoxia and capacity for angiogenesis and inflammation in young diet-sensitive C57 mice compared with diet-resistant FVB mice. Int J Obes (Lond) 2013;37:853-60.ArticlePubMedPDF
- 11. Eldar-Finkelman H, Schreyer SA, Shinohara MM, LeBoeuf RC, Krebs EG. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 1999;48:1662-6.ArticlePubMedPDF
- 12. Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988;37:1163-7.ArticlePubMed
- 13. West DB, Boozer CN, Moody DL, Atkinson RL. Dietary obesity in nine inbred mouse strains. Am J Physiol 1992;262(6 Pt 2):R1025-32.ArticlePubMed
- 14. Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, et al. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism 1995;44:645-51.ArticlePubMed
- 15. Almind K, Kahn CR. Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes 2004;53:3274-85.ArticlePubMedPDF
- 16. Chiu KC, Cohan P, Lee NP, Chuang LM. Insulin sensitivity differs among ethnic groups with a compensatory response in beta-cell function. Diabetes Care 2000;23:1353-8.ArticlePubMedPDF
- 17. Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE; American Diabetes Association GENNID Study Group. Beta-cell function is a major contributor to oral glucose tolerance in high-risk relatives of four ethnic groups in the U.S. Diabetes 2002;51:2170-8.PubMed
- 18. Gerstein HC, Anand S, Yi QL, Vuksan V, Lonn E, Teo K, et al. The relationship between dysglycemia and atherosclerosis in South Asian, Chinese, and European individuals in Canada: a randomly sampled cross-sectional study. Diabetes Care 2003;26:144-9.PubMed
- 19. Davis TM, Mulder H, Lokhnygina Y, Aschner P, Chuang LM, Raffo Grado CA, et al. Effect of race on the glycaemic response to sitagliptin: insights from the Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS). Diabetes Obes Metab 2018;20:1427-34.ArticlePubMedPDF
- 20. Ma RC, Chan JC. Type 2 diabetes in East Asians: similarities and differences with populations in Europe and the United States. Ann N Y Acad Sci 2013;1281:64-91.ArticlePubMedPMCPDF
- 21. Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-Cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 2013;36:111-7.PubMed
- 22. Kou K, Saisho Y, Satoh S, Yamada T, Itoh H. Change in β-cell mass in Japanese nondiabetic obese individuals. J Clin Endocrinol Metab 2013;98:3724-30.ArticlePubMed
- 23. Roh E, Kim KM, Park KS, Kim YJ, Chun EJ, Choi SH, et al. Comparison of pancreatic volume and fat amount linked with glucose homeostasis between healthy Caucasians and Koreans. Diabetes Obes Metab 2018;20:2642-52.ArticlePubMedPDF
- 24. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008;10 Suppl 4:32-42.PubMed
- 25. Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986;1:1077-81.ArticlePubMed
- 26. Simeoni U, Osmond C, Garay R, Buffat C, Boubred F, Chagnaud C, et al. Leptin and insulin in young adulthood areassociated with weight in infancy. J Endocrinol 2020;244:249-59.ArticlePubMed
- 27. Huang RC, Prescott SL, Godfrey KM, Davis EA. Assessment of cardiometabolic risk in children in population studies: underpinning developmental origins of health and disease mother-offspring cohort studies. J Nutr Sci 2015;4:e12.ArticlePubMedPMC
- 28. Heindel JJ, Blumberg B. Environmental obesogens: mechanisms and controversies. Annu Rev Pharmacol Toxicol 2019;59:89-106.ArticlePubMed
- 29. Tain YL, Hsu CN. Developmental programming of the metabolic syndrome: can we reprogram with resveratrol? Int J Mol Sci 2018;19:2584.ArticlePubMedPMC
- 30. Lee WC, Wu KLH, Leu S, Tain YL. Translational insights on developmental origins of metabolic syndrome: focus on fructose consumption. Biomed J 2018;41:96-101.ArticlePubMedPMC
- 31. Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol 2001;185:93-8.ArticlePubMed
- 32. Painter RC, de Rooij SR, Bossuyt PM, de Groot E, Stok WJ, Osmond C, et al. Maternal nutrition during gestation and carotid arterial compliance in the adult offspring: the Dutch famine birth cohort. J Hypertens 2007;25:533-40.ArticlePubMed
- 33. Yura S, Itoh H, Sagawa N, Yamamoto H, Masuzaki H, Nakao K, et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab 2005;1:371-8.ArticlePubMed
- 34. Ozanne SE, Hales CN. Lifespan: catch-up growth and obesity in male mice. Nature 2004;427:411-2.ArticlePDF
- 35. Stettler N, Zemel BS, Kumanyika S, Stallings VA. Infant weight gain and childhood overweight status in a multicenter, cohort study. Pediatrics 2002;109:194-9.ArticlePubMedPDF
- 36. Nordman H, Jaaskelainen J, Voutilainen R. Birth size as a determinant of cardiometabolic risk factors in children. Horm Res Paediatr 2020;93:144-53.ArticlePubMedPDF
- 37. Shi H, Yang X, Wu D, Wang X, Li T, Liu H, et al. Insights into infancy weight gain patterns for term small-for-gestationalage babies. Nutr J 2018;17:97.ArticlePubMedPMCPDF
- 38. Berends LM, Dearden L, Tung YC, Voshol P, Fernandez-Twinn DS, Ozanne SE. Programming of central and peripheral insulin resistance by low birthweight and postnatal catchup growth in male mice. Diabetologia 2018;61:2225-34.ArticlePubMedPMCPDF
- 39. Yoshida Y, Fuchita M, Kimura-Koyanagi M, Kanno A, Matsuda T, Asahara SI, et al. Contribution of insulin signaling to the regulation of pancreatic beta-cell mass during the catch-up growth period in a low birth weight mouse model. Diabetol Int 2014;5:43-52.ArticlePDF
- 40. Inoue T, Kido Y, Asahara S, Matsuda T, Shibutani Y, Koyanagi M, et al. Effect of intrauterine undernutrition during late gestation on pancreatic beta cell mass. Biomed Res 2009;30:325-30.ArticlePubMed
- 41. Stewart CP, Christian P, Schulze KJ, Arguello M, LeClerq SC, Khatry SK, et al. Low maternal vitamin B-12 status is associated with offspring insulin resistance regardless of antenatal micronutrient supplementation in rural Nepal. J Nutr 2011;141:1912-7.ArticlePubMed
- 42. Ozanne SE, Olsen GS, Hansen LL, Tingey KJ, Nave BT, Wang CL, et al. Early growth restriction leads to down regulation of protein kinase C zeta and insulin resistance in skeletal muscle. J Endocrinol 2003;177:235-41.ArticlePubMed
- 43. Ozanne SE, Jensen CB, Tingey KJ, Storgaard H, Madsbad S, Vaag AA. Low birthweight is associated with specific changes in muscle insulin-signalling protein expression. Diabetologia 2005;48:547-52.ArticlePubMedPDF
- 44. McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009;119:323-35.ArticlePubMedPMC
- 45. Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 2010;467:963-6.ArticlePubMedPDF
- 46. Gruenert DC, Cozens AL. Inheritance of phenotype in mammalian cells: genetic vs. epigenetic mechanisms. Am J Physiol 1991;260(6 Pt 1):L386-94.ArticlePubMed
- 47. Takeshima H, Ushijima T. Accumulation of genetic and epigenetic alterations in normal cells and cancer risk. NPJ Precis Oncol 2019;3:7.ArticlePubMedPMCPDF
- 48. Dalfra MG, Burlina S, Del Vescovo GG, Lapolla A. Genetics and epigenetics: new insight on gestational diabetes mellitus. Front Endocrinol (Lausanne) 2020;11:602477.PubMedPMC
- 49. Duran Fernandez-Feijoo C, Carrasco Carrasco C, Villalmazo Francisco N, Cebria Romero J, Fernandez Lorenzo JR, Jimenez-Chillaron JC, et al. Influence of catch up growth on spatial learning and memory in a mouse model of intrauterine growth restriction. PLoS One 2017;12:e0177468.ArticlePubMedPMC
- 50. Suter MA, Ma J, Vuguin PM, Hartil K, Fiallo A, Harris RA, et al. In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am J Obstet Gynecol 2014;210:463.ArticlePubMedPMC
- 51. Tozour J, Hughes F, Carrier A, Vieau D, Delahaye F. Prenatal hyperglycemia exposure and cellular stress, a sugar-coated view of early programming of metabolic diseases. Biomolecules 2020;10:1359.ArticlePubMedPMC
- 52. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 2008;118:2316-24.ArticlePubMedPMC
- 53. Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, UribeLewis S, Ito Y, et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc Natl Acad Sci U S A 2011;108:5449-54.ArticlePubMedPMC
- 54. Einstein F, Thompson RF, Bhagat TD, Fazzari MJ, Verma A, Barzilai N, et al. Cytosine methylation dysregulation in neonates following intrauterine growth restriction. PLoS One 2010;5:e8887.ArticlePubMedPMC
- 55. Xing Y, Zhang J, Wei H, Zhang H, Guan Y, Wang X, et al. Reduction of the PI3K/Akt related signaling activities in skeletal muscle tissues involves insulin resistance in intrauterine growth restriction rats with catch-up growth. PLoS One 2019;14:e0216665.ArticlePubMedPMC
- 56. Dunlop K, Cedrone M, Staples JF, Regnault TR. Altered fetal skeletal muscle nutrient metabolism following an adverse in utero environment and the modulation of later life insulin sensitivity. Nutrients 2015;7:1202-16.ArticlePubMedPMC
- 57. Raychaudhuri N, Raychaudhuri S, Thamotharan M, Devaskar SU. Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J Biol Chem 2008;283:13611-26.ArticlePubMedPMC
- 58. Ehara T, Kamei Y, Takahashi M, Yuan X, Kanai S, Tamura E, et al. Role of DNA methylation in the regulation of lipogenic glycerol-3-phosphate acyltransferase 1 gene expression in the mouse neonatal liver. Diabetes 2012;61:2442-50.ArticlePubMedPMCPDF
- 59. Martinez D, Pentinat T, Ribo S, Daviaud C, Bloks VW, Cebria J, et al. In utero undernutrition in male mice programs liver lipid metabolism in the second-generation offspring involving altered Lxra DNA methylation. Cell Metab 2014;19:941-51.ArticlePubMed
- 60. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet 2014;383:69-82.ArticlePubMed
- 61. Incani M, Serafini C, Satta C, Perra L, Scano F, Frongia P, et al. High prevalence of diabetes-specific autoimmunity in firstdegree relatives of Sardinian patients with type 1 diabetes. Diabetes Metab Res Rev 2017;33:e2864.ArticlePDF
- 62. You WP, Henneberg M. Type 1 diabetes prevalence increasing globally and regionally: the role of natural selection and life expectancy at birth. BMJ Open Diabetes Res Care 2016;4:e000161.ArticlePubMedPMC
- 63. Monaghan M, Helgeson V, Wiebe D. Type 1 diabetes in young adulthood. Curr Diabetes Rev 2015;11:239-50.ArticlePubMedPMC
- 64. DIAMOND Project Group. Incidence and trends of childhood type 1 diabetes worldwide 1990-1999. Diabet Med 2006;23:857-66.ArticlePubMed
- 65. Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G; EURODIAB Study Group. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet 2009;373:2027-33.ArticlePubMed
- 66. Noble JA, Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diab Rep 2011;11:533-42.ArticlePubMedPMCPDF
- 67. Mishra R, Akerlund M, Cousminer DL, Ahlqvist E, Bradfield JP, Chesi A, et al. Genetic discrimination between LADA and childhood-onset type 1 diabetes within the MHC. Diabetes Care 2020;43:418-25.ArticlePubMedPDF
- 68. Buzzetti R, Prudente S, Copetti M, Dauriz M, Zampetti S, Garofolo M, et al. Clinical worthlessness of genetic prediction of common forms of diabetes mellitus and related chronic complications: a position statement of the Italian Society of Diabetology. Nutr Metab Cardiovasc Dis 2017;27:99-114.PubMed
- 69. NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016;387:1377-96.ArticlePubMedPMC
- 70. Asahara SI, Miura H, Ogawa W, Tamori Y. Sex difference in the association of obesity with personal or social background among urban residents in Japan. PLoS One 2020;15:e0242105.ArticlePubMedPMC
- 71. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 2017;390:2627-42.PubMedPMC
- 72. De Keukelaere M, Fieuws S, Reynaert N, Vandoorne E, Kerckhove KV, Asscherickx W, et al. Evolution of body mass index in children with type 1 diabetes mellitus. Eur J Pediatr 2018;177:1661-6.ArticlePubMedPDF
- 73. Corbin KD, Driscoll KA, Pratley RE, Smith SR, Maahs DM, Mayer-Davis EJ, et al. Obesity in type 1 diabetes: pathophysiology, clinical impact, and mechanisms. Endocr Rev 2018;39:629-63.ArticlePubMed
- 74. Patterson CC, Harjutsalo V, Rosenbauer J, Neu A, Cinek O, Skrivarhaug T, et al. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 1989-2013: a multicentre prospective registration study. Diabetologia 2019;62:408-17.ArticlePubMedPDF
- 75. Public Health Report: Overweight and obesity in Norway. Available from: https://www.fhi.no/en/op/hin/health-disease/overweight-and-obesity-in-norway--- (updated 2017 Nov 3).
- 76. Fellinger P, Fuchs D, Wolf P, Heinze G, Luger A, Krebs M, et al. Overweight and obesity in type 1 diabetes equal those of the general population. Wien Klin Wochenschr 2019;131:55-60.ArticlePubMedPMCPDF
- 77. Ferrara CT, Geyer SM, Liu YF, Evans-Molina C, Libman IM, Besser R, et al. Excess BMI in childhood: a modifiable risk factor for type 1 diabetes development? Diabetes Care 2017;40:698-701.ArticlePubMedPMCPDF
- 78. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest 2017;127:1-4.ArticlePubMedPMC
- 79. Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet 2016;387:2340-8.ArticlePubMedPMC
- 80. Nekoua MP, Bertin A, Sane F, Alidjinou EK, Lobert D, Trauet J, et al. Pancreatic beta cells persistently infected with coxsackievirus B4 are targets of NK cell-mediated cytolytic activity. Cell Mol Life Sci 2020;77:179-94.ArticlePubMedPDF
- 81. Lamb MM, Miller M, Seifert JA, Frederiksen B, Kroehl M, Rewers M, et al. The effect of childhood cow’s milk intake and HLA-DR genotype on risk of islet autoimmunity and type 1 diabetes: the Diabetes Autoimmunity Study in the Young. Pediatr Diabetes 2015;16:31-8.ArticlePubMed
- 82. Lempainen J, Tauriainen S, Vaarala O, Makela M, Honkanen H, Marttila J, et al. Interaction of enterovirus infection and cow’s milk-based formula nutrition in type 1 diabetes-associated autoimmunity. Diabetes Metab Res Rev 2012;28:177-85.ArticlePubMed
- 83. Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA 2003;290:1721-8.ArticlePubMed
- 84. Uusitalo U, Lee HS, Andren Aronsson C, Vehik K, Yang J, Hummel S, et al. Early infant diet and islet autoimmunity in the TEDDY study. Diabetes Care 2018;41:522-30.PubMedPMC
- 85. Op de Beeck A, Eizirik DL. Viral infections in type 1 diabetes mellitus: why the β cells? Nat Rev Endocrinol 2016;12:263-73.ArticlePubMedPMCPDF
- 86. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013;56:234-41.ArticlePubMedPDF
- 87. Thomaidou S, Zaldumbide A, Roep BO. Islet stress, degradation and autoimmunity. Diabetes Obes Metab 2018;20(Suppl 2):88-94.ArticlePubMedPMCPDF
- 88. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med 2017;23:501-7.ArticlePubMedPDF
- 89. Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008;40:1092-7.ArticlePubMedPDF
- 90. Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 2008;40:1098-102.ArticlePubMedPDF
- 91. Hu C, Wang C, Zhang R, Ma X, Wang J, Lu J, et al. Variations in KCNQ1 are associated with type 2 diabetes and beta cell function in a Chinese population. Diabetologia 2009;52:1322-5.ArticlePubMedPDF
- 92. Jonsson A, Isomaa B, Tuomi T, Taneera J, Salehi A, Nilsson P, et al. A variant in the KCNQ1 gene predicts future type 2 diabetes and mediates impaired insulin secretion. Diabetes 2009;58:2409-13.ArticlePubMedPMCPDF
- 93. Tan JT, Nurbaya S, Gardner D, Ye S, Tai ES, Ng DP. Genetic variation in KCNQ1 associates with fasting glucose and betacell function: a study of 3,734 subjects comprising three ethnicities living in Singapore. Diabetes 2009;58:1445-9.PubMedPMC
- 94. Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes 2012;61:1726-33.ArticlePubMedPMCPDF
- 95. Asahara S, Etoh H, Inoue H, Teruyama K, Shibutani Y, Ihara Y, et al. Paternal allelic mutation at the Kcnq1 locus reduces pancreatic β-cell mass by epigenetic modification of Cdkn1c. Proc Natl Acad Sci U S A 2015;112:8332-7.ArticlePubMedPMC
- 96. Matsuda T, Kido Y, Asahara S, Kaisho T, Tanaka T, Hashimoto N, et al. Ablation of C/EBPbeta alleviates ER stress and pancreatic beta cell failure through the GRP78 chaperone in mice. J Clin Invest 2010;120:115-26.ArticlePubMed
- 97. Matsuda T, Takahashi H, Mieda Y, Shimizu S, Kawamoto T, Matsuura Y, et al. Regulation of pancreatic β cell mass by cross-interaction between CCAAT enhancer binding protein β induced by endoplasmic reticulum stress and AMP-activated protein kinase activity. PLoS One 2015;10:e0130757.ArticlePubMedPMC
- 98. Deval C, Chaveroux C, Maurin AC, Cherasse Y, Parry L, Carraro V, et al. Amino acid limitation regulates the expression of genes involved in several specific biological processes through GCN2-dependent and GCN2-independent pathways. FEBS J 2009;276:707-18.ArticlePubMed
- 99. Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 2002;22:6681-8.PubMedPMC
- 100. Guo F, Cavener DR. The GCN2 eIF2alpha kinase regulates fatty-acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metab 2007;5:103-14.PubMed
- 101. Miyake K, Yang W, Hara K, Yasuda K, Horikawa Y, Osawa H, et al. Construction of a prediction model for type 2 diabetes mellitus in the Japanese population based on 11 genes with strong evidence of the association. J Hum Genet 2009;54:236-41.ArticlePubMedPDF
- 102. Kanno A, Asahara SI, Furubayashi A, Masuda K, Yoshitomi R, Suzuki E, et al. GCN2 regulates pancreatic β cell mass by sensing intracellular amino acid levels. JCI Insight 2020;5:e128820.ArticlePubMedPMC
- 103. Kanno A, Asahara SI, Masuda K, Matsuda T, Kimura-Koyanagi M, Seino S, et al. Compensatory hyperinsulinemia in high-fat diet-induced obese mice is associated with enhanced insulin translation in islets. Biochem Biophys Res Commun 2015;458:681-6.ArticlePubMed
- 104. Zhou T, Kim TW, Chong CN, Tan L, Amin S, Sadat Badieyan Z, et al. A hPSC-based platform to discover gene-environment interactions that impact human β-cell and dopamine neuron survival. Nat Commun 2018;9:4815.ArticlePubMedPMCPDF
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Increased risk of incident diabetes after therapy with immune checkpoint inhibitor compared with conventional chemotherapy: A longitudinal trajectory analysis using a tertiary care hospital database
Minyoung Lee, Kyeongseob Jeong, Yu Rang Park, Yumie Rhee
Metabolism.2023; 138: 155311. CrossRef - The ameliorating effects of mesenchymal stem cells compared to α‐tocopherol on apoptosis and autophagy in streptozotocin‐induced diabetic rats: Implication of PI3K/Akt signaling pathway and entero‐insular axis
Heba A. Mubarak, Manal M. Kamal, Yossra Mahmoud, Fatma S. Abd‐Elsamea, Eman Abdelbary, Marwa G. Gamea, Reham I. El‐Mahdy
Journal of Cellular Biochemistry.2023; 124(11): 1705. CrossRef - Leptin Rs7799039 polymorphism is associated with type 2 diabetes mellitus Egyptian patients
Amal Ahmed Mohamed, Dina M. Abo-Elmatty, Alaa S. Wahba, Omnia Ezzat Esmail, Hadeer Saied Mahmoud Salim, Wafaa Salah Mohammed Hegab, Mona Mostafa Farid Ghanem, Nadia Youssef Riad, Doaa Ghaith, Lamiaa I Daker, Shorouk Issa, Noha Hassan Radwan, Eman Sultan,
Archives of Physiology and Biochemistry.2023; : 1. CrossRef - Association of Polygenic Variants with Type 2 Diabetes Risk and Their Interaction with Lifestyles in Asians
Haeng Jeon Hur, Hye Jeong Yang, Min Jung Kim, Kyun-Hee Lee, Myung-Sunny Kim, Sunmin Park
Nutrients.2022; 14(15): 3222. CrossRef - Chemical Compounds and Ambient Factors Affecting Pancreatic Alpha-Cells Mass and Function: What Evidence?
Gaia Chiara Mannino, Elettra Mancuso, Stefano Sbrignadello, Micaela Morettini, Francesco Andreozzi, Andrea Tura
International Journal of Environmental Research and Public Health.2022; 19(24): 16489. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite