- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 43(3); 2019 > Article
-
ReviewObesity and Metabolic Syndrome Understanding Bile Acid Signaling in Diabetes: From Pathophysiology to Therapeutic Targets
-
Jessica M. Ferrell
 , John Y. L. Chiang
, John Y. L. Chiang -
Diabetes & Metabolism Journal 2019;43(3):257-272.
DOI: https://doi.org/10.4093/dmj.2019.0043
Published online: June 13, 2019
Integrative Medical Sciences, Northeast Ohio Medical University, Rootstown, OH, USA.
- Corresponding author: John Y. L. Chiang. Integrative Medical Sciences Northeast Ohio Medical University, SR44, PO Box 95 Rootstown, OH 44272, USA. jchiang@neomed.edu
• Received: March 6, 2019 • Accepted: April 25, 2019
Copyright © 2019 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- BILE ACID BIOLOGY AND PHYSIOLOGY
- BILE ACID SIGNALING IN METABOLIC REGULATION
- BILE ACID SIGNALING IN DIABETES
- INTERACTIONS BETWEEN BILE ACIDS AND THE GUT MICROBIOTA
- METABOLIC AND BARIATRIC SURGERY IMPROVES OBESITY AND DIABETES
- BILE ACID-BASED THERAPY FOR DIABETES
- CONCLUSIONS
- ACKNOWLEDGMENTS
- NOTES
- REFERENCES
ABSTRACT
- Diabetes and obesity have reached an epidemic status worldwide. Diabetes increases the risk for cardiovascular disease and non-alcoholic fatty liver disease. Primary bile acids are synthesized in hepatocytes and are transformed to secondary bile acids in the intestine by gut bacteria. Bile acids are nutrient sensors and metabolic integrators that regulate lipid, glucose, and energy homeostasis by activating nuclear farnesoid X receptor and membrane Takeda G protein-coupled receptor 5. Bile acids control gut bacteria overgrowth, species population, and protect the integrity of the intestinal barrier. Gut bacteria, in turn, control circulating bile acid composition and pool size. Dysregulation of bile acid homeostasis and dysbiosis causes diabetes and obesity. Targeting bile acid signaling and the gut microbiome have therapeutic potential for treating diabetes, obesity, and non-alcoholic fatty liver disease.
- The global epidemic of obesity has caused the increase of the prevalence of type 2 diabetes mellitus (T2DM), which is predicted to increase 54% by 2030 [1]. T2DM is a complex metabolic disease developed most often in middle-aged and older people with a family history of diabetes and obesity. Asian, Hispanic, American Indian, and African American populations have high prevalence of T2DM. The majority of diabetes patients (70%) are overweight or obese, and the obesity rate is expected to increase 33% in the next 20 years [2]. Diabetic and obese patients have increased risk for cardiovascular disease (CVD), the leading cause of death in western countries (Fig. 1). T2DM patients are insulin resistant and glucose intolerant in skeletal muscle, adipose tissue, and liver. Consequently, hyperglycemia induces vascular complications in blood vessels, kidney, heart, and liver, and can cause renal and retinal disorders and macrovascular complications, leading to blindness, kidney failure, atherosclerosis, stroke, and liver diseases.
- T2DM is linked to non-alcoholic fatty liver disease (NAFLD) [3], which has rapidly increased worldwide and has a global prevalence of about 24%; the highest rates being in South America, the Middle East, Asia, the USA, and Europe [4]. NAFLD is a significant complication of obesity and diabetes [5] and is an independent risk factor for CVD (Fig. 1) [678]. CVD and NAFLD are the heart and liver manifestations of the metabolic syndrome (syndrome X) [9], a collection of five abnormal metabolic phenotypes: hypertension, hyperglycemia, hypertriglyceridemia, insulin resistance, and obesity (Fig. 1) [10]. NAFLD consists of a broad spectrum of liver diseases from simple hepatic steatosis and nonalcoholic steatohepatitis (NASH) to cirrhosis and hepatocellular carcinoma (HCC), the end stages of liver disease [111213]. High fat, high carbohydrate, or high calorie diets, alcohol, starvation, drugs, and viral infection can cause hepatic steatosis [14]. Simple steatosis is reversible, about 30% of NAFLD patients progress to NASH, a chronic condition with liver inflammation, macrovascular ballooning, macrophage infiltration and fibrosis. About 2% to 5% of NASH patients develop liver cirrhosis and HCC. NASH has become the second leading cause of liver cancer and will surpass viral hepatitis as the primary cause for liver transplants. Multiple factors, including reactive oxidizing species, visceral fat, viruses, cigarette smoking, insulin resistance, lipotoxicity, and hepatic cholesterol have been linked to the progression of simple hepatic steatosis to NASH [15], and there is no U.S. Food and Drug Administration-approved therapeutic drug for treating NASH.
- Bile acids are derived from cholesterol catabolism in the liver and are amphipathic molecules with strong detergent properties that aid in the absorption of dietary fats and steroids, lipid-soluble vitamins, and xenobiotics, including drugs and environmental contaminates. Bile acids are now recognized as key endogenous steroid molecules that play critical roles in regulating and maintaining lipid, glucose and energy metabolism, protecting against inflammation in the liver, intestine and heart, and preventing diabetes and obesity [16]. Bile acid metabolism is altered in T2DM [171819] and dysregulation of lipid, glucose and energy metabolism causes inflammatory metabolic diseases including T2DM, NAFLD, and CVD. This review will cover bile acid pathophysiology and signaling, recent advances in understanding the role of bile acid signaling in diabetes, and bile acids as drug therapies for treating metabolic diseases.
INTRODUCTION
- Bile acid biology
- Bile acids are the end products of cholesterol catabolism in the liver, the only organ which has all the enzymes required in the cascade pathway to convert excessive cholesterol to bile acids. Bile acid synthesis accounts for catabolism of about 50% of the daily cholesterol output. Biliary secretion of cholesterol in bile accounts for another 40% of the daily cholesterol output. The remaining 10% of cholesterol is utilized for membrane synthesis and steroid hormone synthesis in steroidogenic tissues. Therefore, bile acid metabolism plays a critical role in maintaining whole body cholesterol homeostasis.
- Bile acid synthesis in the liver
- In the human liver, cholesterol 7α-hydroxylase (CYP7A1) catalyzes the first and rate-limiting step in the classic bile acid synthesis pathway and synthesizes two primary bile acids, chenodeoxycholic acid (CDCA) and cholic acid (CA), the latter of which requires sterol 12α-hydroxylase (CYP8B1) (Fig. 2). CA has 3 hydroxyl (HO) groups at the 3α, 7α, and 12α-positions; thus, it is more soluble (hydrophilic) than CDCA, which has 2 hydroxyl groups at the 3α and 7α-positions. All hydroxyl groups in bile acids are facing one side of the carbon skeleton, creating a hydrophilic side and a hydrophobic side, making bile acids amphipathic molecules with strong detergent properties. Mitochondrial sterol 27-hydroxylase (CYP27A1) then catalyzes steroid side-chain oxidation, followed by a peroxisomal β-oxidation reaction that cleaves a 3-carbon unit from the steroid-side chain to form C-24 bile acids and propionyl-CoA. Bile acids also can be synthesized via the alternative pathway, initiated by CYP27A1, to form 27-hydroxycholesterol and 3β-hydroxy-5-cholestenoic acid, which is then hydroxylated at the 7α-position by the non-specific oxysterol 7α-hydroxylase (CYP7B1). CYP27A1 and CYP7B1 are expressed in most tissues and macrophages and are responsible for the metabolism of oxysterols to steroid hormones in steroidogenic tissues; these oxidized steroid intermediates can be transported to the liver for synthesis of bile acids. The classic pathway is the major route for bile acid synthesis in humans. In rodents, most CDCA (3α, 7α) is converted to α-muricholic acid (α-MCA, 3α, 6β, 7α) and the 7α-HO group is epimerized to 7β-HO by Cyp2c70 (sterol 6β-hydroxylase), forming β-muricholic acid (β-MCA, 3α, 6β, 7β). Addition of a 6β-HO group to CDCA converts hydrophobic CDCA to highly soluble and non-toxic α-MCA and β-MCA. In contrast to humans, CA (50%) and α-MCA plus β-MCAs (50%) are the predominant primary bile acids produced in mouse liver [20].
- Most bile acids are conjugated to glycine (G) and taurine (T) in a ratio of about 3:1 in humans. In mice, most bile acids (>95%) are taurine-conjugated. The conjugated bile acids are secreted into bile and stored in the gallbladder, and after meal intake, bile acids are secreted into the intestinal tract. Bile acids are reabsorbed, mostly in the terminal ileum and colon, and are secreted into portal blood circulation back to the liver to inhibit bile acid synthesis. This enterohepatic circulation of bile acids from the liver to intestine and back to the liver occurs six to eight times a day and is highly efficient in reabsorbing about 95% of bile acids in a pool of about 10 g in an average human. Small amounts of bile acids lost in feces (5%, 0.5 g/day) are replenished by de novo synthesis in the liver (Fig. 2) [20].
- Bile acid biotransformation in the gut
- The gut bacteria metabolize primary bile acids to secondary bile acids, which were once considered “damaged” bile acids that were excreted into feces or cleared in urine. In the intestine, a portion of conjugated CA and CDCA are de-conjugated by gut bacterial bile salt hydroxylase (BSH) to free bile acids, then bacterial 7α-dehydroxylase activity removes a 7-HO group from CA and CDCA to form deoxycholic acid (DCA) and lithocholic acid (LCA), respectively (Fig. 2) [21]. LCA is a toxic and highly insoluble bile acid, most of which is excreted into feces, though small amounts of LCA (approximately 2%) are circulated to the liver and sulfoconjugated for secretion into urine. DCA is a potent bactericide that controls bacterial overgrowth, but also is a promoter of colon cancer. Small amounts of CDCA (1% to 2%) are converted to its 7β-epimer, ursodeoxycholic acid (UDCA) by gut bacterial 7β-hydroxysteroid dehydrogenase in humans. Epimerization of the C7-HO group from the α- to the β-position converts toxic CDCA to hydrophilic and non-toxic UDCA. In humans, the circulating bile acid pool is highly hydrophobic, consisting of CA, CDCA, and DCA in a ratio of about 40:40:20, and the ratio of glycine to taurine-conjugated bile acids is about 3 to 1 [20].
BILE ACID BIOLOGY AND PHYSIOLOGY
- Extensive research in the last three decades has identified bile acids as signaling molecules that activate several nuclear receptors: farnesoid X receptor (FXR) [222324], vitamin D receptor (VDR) [25], pregnane X receptor (PXR) [26]; and the membrane G protein-coupled receptors: Takeda G protein-coupled receptor 5 (TGR5) [27], sphingosine-1 phosphate receptor 2 (S1PR2) [28], and muscarinic M2 receptor [29]. These bile acid-activated receptors play critical roles in liver metabolism [30]. This section will focus on the roles of FXR and TGR5 in the regulation of metabolism and pathophysiology of liver-related metabolic diseases.
- Farnesoid X receptor
- FXR is mainly expressed in the digestive system, including liver and intestine. FXR is activated by bile acids in the order of potency CDCA>LCA=DCA>CA. FXR knockout mice have increased hepatic triglycerides, cholesterol and a proatherogenic lipid profile, and reduced bile acid pool and increased fecal bile acid secretion, indicating FXR plays a major role in bile acid and lipid metabolism [31]. FXR also regulates the enterohepatic circulation of bile acids and feedback homeostasis [32]. In the liver, bile acids activate FXR to induce the expression of the major hepatic bile acid efflux transporter, bile salt export pump which secretes conjugated bile acids into bile, and inhibits the sinusoidal hepatic bile acid uptake transporter, Na+2-dependent taurocholate co-transport peptide (Fig. 2). These two major bile acid transporters regulate hepatic bile acid homeostasis. In hepatocytes, bile acid activation of FXR induces a transcriptional repressor, small heterodimer partner, to inhibit transcription of the CYP7A1 and CYP8B1 genes (Fig. 2). In the ileum, bile acids are reabsorbed into enterocytes via apical sodium-dependent bile acid transporter, whose function is inhibited by bile acids. Bile acids activate intestinal FXR to induce the release of the intestinal hormone fibroblast growth factor 19 (FGF19) in humans or FGF15 in mice (Fig. 2). FXR also induces the bile acid efflux transporters organic solute transporter α and β (OSTα/OSTβ) to secrete bile acids into portal blood circulation. FGF19 released from enterocytes is transported via portal blood circulation to hepatocytes and binds to the membrane FGF receptor 4/β-Klotho complex, which activates mitogen-activated protein kinase (MAPK) and extracellular regulated kinase 1 and 2 (ERK1/2) signaling to inhibit CYP7A1 and CYP8B1 gene transcription (Fig. 2). The intestinal FXR/FGF19 to hepatic FGFR4 pathway may be the major physiological mechanism for bile acid feedback regulation of bile acid synthesis.
- The role of FXR in glucose metabolism is controversial. It was reported that FXR expression was reduced in streptozotocin-induced diabetic rats, and insulin and glucose induced FXR expression [33]. Activation of FXR has been shown to improve glucose and lipid metabolism and reduce inflammation in diabetes [343536]. Surprisingly, other studies reported that Fxr−/− mice had improved hyperglycemia and insulin sensitivity, and activation of FXR induced obesity and diabetes by reducing energy expenditure [37]. In pancreatic β-cells, activation of FXR stimulates glycolysis to increase the adenosine triphosphate:adenosine diphosphate (ATP:ADP) ratio and results in closing KATP-channels and depolarizing the plasma membrane, which subsequently opens Ca2+ channels, increasing Ca2+ influx and stimulating insulin secretion from β-cells [38].
- Takeda G protein-coupled receptor 5
- In the liver, TGR5 is expressed in sinusoidal endothelial cells, Kupffer cells (hepatic resident macrophages), stellate cells, and biliary epithelial cells in bile ducts, but not in hepatocytes [394041], and the secondary bile acids LCA and DCA are potent endogenous TGR5 agonists (LCA>DCA>CDCA>CA). TGR5 is expressed in the epithelium of human gallbladder and controls gallbladder refiling [42]. TGR5 also plays a key role in bile acid metabolism and fasting-induced hepatic steatosis [43]. In the colon, TGR5 mediates bile acid-induced gastrointestinal motility, transit time and defecation [44]. In the intestine and macrophage, activation of TGR5 protects against inflammation [45]. TGR5 also plays a critical role in the control of glucose homeostasis [46]. Activation of TGR5 stimulates the release of glucagon-like peptide-1 (GLP-1) from enteroendocrine L-cells to stimulate insulin secretion from β-cells and increase insulin sensitivity. In adipose tissue, activation of TGR5 induces thyroid hormone deiodinase type 2 (DIO2), which converts thyroid hormone thyroxine (T4) to triiodothyronine (T3) to stimulate energy metabolism and white adipose tissue browning (Fig. 2). CDCA increases brown adipose tissue in humans, likely through TGR5-mediated increase of uncoupling protein and DIO2 expression [47]. TGR5 knockout mice are protected from cholesterol gallstone disease and high fat diet (HFD) induced obesity [4849]. FXR and TGR5 are co-expressed in L-cells, and activation of intestinal FXR stimulates TGR5 gene transcription via an FXR response element located in the TGR5 gene promoter; this crosstalk stimulates GLP-1 secretion [50]. It is therefore likely that some of the reported FXR effects on glucose metabolism may be due to TGR5 signaling in the gut.
BILE ACID SIGNALING IN METABOLIC REGULATION
- Bile acids are nutrient sensors and metabolic regulators
- Bile acid synthesis and CYP7A1 expression exhibit circadian rhythms, which are modulated by fasting and feeding. Sleep disruption, alcohol, and HFD disrupt these rhythms and cause altered bile acid homeostasis, contributing to the pathogenesis of insulin resistance and obesity [2051525354]. Bile acids are metabolic sensors that aid in dietary nutrient absorption to ultimately provide fuel for energy metabolism and biosynthesis.
- Post-prandial regulation
- Feeding rapidly stimulates the release of bile acids stored in the gallbladder and de-represses CYP7A1 expression to stimulate bile acid synthesis during the postprandial state. Feeding inhibits CYP8B1 expression, reducing CA and increasing CDCA, which is a more potent FXR agonist than CA. In the postprandial state, bile acids activate FXR to inhibit hepatic lipogenesis by stimulating insulin and insulin receptor substrate 1 (IRS1)-AKT-phosphoinositide 3-kinase (PI3K) signaling, which inhibits mechanistic target of rapamycin complex 1 (mTORC1) and induces autophagy (Fig. 3) [5556]. mTORC1-pS6K promotes maturation and nuclear localization of steroid regulatory element binding protein 1c (SREBP1c) to stimulate lipogenesis [57]. FXR inhibition of the mTORC1-SREBP1c pathway may be the most plausible mechanism for bile acid inhibition of lipogenesis.
- Post-absorptive regulation
- During the late postprandial state or post-absorptive, intestinal FXR induces human FGF19 production and secretion from enterocytes to regulate bile acid synthesis in the liver via FGFR4/βKlotho/ERK1/2 signaling [58], which inhibits CYP7A1 and bile acid synthesis [59].
- Fasting and starvation
- During fasting, serum insulin and glucose levels decrease while glucagon increases to induce gluconeogenesis and release glucose from glycogen to prevent hypoglycemia. Glucagon stimulates hormone sensitive lipase A to release free fatty acids from triglycerides in adipose tissue (Fig. 3). During prolonged fasting and starvation, free fatty acids activate peroxisome proliferator-activated receptor α (PPARα) to induce FGF21 and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) in liver and adipose tissue [6061]. FGF21 is a nutrient sensor that stimulates insulin sensitivity and regulates glucose and energy metabolism in adipose tissue. It also reduces serum triglycerides in diet-induced obese mice [60626364] and inhibits mTORC1 signaling in hepatocytes (Fig. 3) [63]. Fasting induces CYP8B1, resulting in increased CA and DCA in the bile acid pool, which induces ceramide synthesis via FXR and activates hepatic mTORC1 signaling to stimulate lipogenesis and cause hepatic insulin resistance (Fig. 3) [65].
- Hyperglycemia
- In hyperglycemia, glucose is converted to acetyl-CoA, which stimulates histone acetylation. Glucose and insulin are known to stimulate CYP7A1 gene transcription by increasing histone acetylation of CYP7A1 chromatin [6667]. This effect is referred to “glucose memory”. In diabetic and obese patients, stimulation of bile acid synthesis increases serum bile acids, with a higher ratio of 12α-hydroxylated bile acids (CA and DCA) to non-12α-hydroxylated bile acids (CDCA) [1819]. Increased CYP8B1 and CA stimulates dietary fat and cholesterol absorption and may contribute to dyslipidemia, diabetes, and obesity. Inhibition of CA synthesis improves glucose homeostasis and prevents diet-induced obesity and atherosclerosis in mouse models [686970].
BILE ACID SIGNALING IN DIABETES
- Bile acids in the gut-to-liver axis
- The gut-to-liver axis plays a critical role in the regulation of hepatic metabolism via the interactions between bile acids and the gut microbiota [717273]. Bile acid biotransformation by gut bacteria determines bile acid composition in the circulating bile acid pool and total bile acid pool size. Bile acids control gut bacterial growth, gut microbial composition, gut barrier function, and production of bacterial metabolites. Interactions between bile acids and gut bacteria significantly impact the health of the host and contribute to the pathogenesis of metabolic diseases, i.e., liver disease, obesity, and diabetes [74]. Dietary factors affect gut microbial growth and metabolism depending on protein, saturated or unsaturated fat, fiber, cholesterol, sucrose, fructose, and carbohydrate content [75]. Diet shapes the gut microbiota to alter host energy metabolism and contributes to diabetes and obesity. HFDs can cause gut dysbiosis and impair intestinal barrier function (leaky gut) by altering the gut microbiome. Gut microbes utilize short-chain fatty acids, mostly acetate, butyrate and propionate, for energy metabolism. Butyrate improves insulin sensitivity and energy metabolism [76] and Chinese T2DM patients had decreased butyrate-producing bacteria compared to healthy control subjects [77].
- The gut microbiota in diabetes
- The human gut microbiome consists of 3 trillion microorganisms in four major phyla, Firmicutes (60%), Bacteroidetes (22%), Actinobacteria (17%), and Proteobacteria (1%) [74]. A high ratio of Firmicutes to Bacteroidetes enables the gut microbiota to extract energy more efficiently from HFDs, increasing adiposity and obesity in humans. Animal-based diets increase the abundance of the bile-tolerant bacteria Bilophila wadsworthia and decreases Firmicutes [78]. High saturated fat diets or low fat diets supplemented with taurocholic acid (TCA) increase B. wadsworthia to promote a proinflammatory response and inflammatory bowel disease [79]. Increased abundance of Lactobacillus is associated with T2DM in humans [77] and in type 1 diabetes mellitus (T1DM) patients, Bacteroidetes increases and Firmicutes decreases. Higher Bacteroides species abundance is associated with autoimmunity, with increase of B. ovatus and B. uniformis, and decrease of B. fragilis in T1DM patients [75]. Interestingly, mucin-degrading Akkermansia muciniphila improves glucose tolerance in T1DM and HFD-fed mice [75].
- Intestinal FXR in diabetes
- T-α-MCA and T-β-MCA have been identified as antagonists of intestinal FXR that reduce bile acid feedback regulation and increase bile acid synthesis [71]. In germ-free mice, bile acid pool size is increased, with increased T-α-MCA and T-β-MCA and reduced TCA. Cyp8b1 deficiency also increased the bile acid pool in mice, with higher T-α-MCA and T-β-MCA, which antagonize intestinal FXR. These mice also had decreased hepatic lipogenesis, improved insulin tolerance, and altered gut microbiota [8081]. Intestinal FXR plays a critical role in metabolic disease via modulation of the microbiome [82]. Antibiotics, the antioxidant tempol, the FXR antagonist Gly-MCA, and deficiency of intestinal FXR all increase conjugated bile acids and T-β-MCAs, and suggest that inactivating intestinal FXR signaling decreases hepatic triglycerides in HFD-fed mice [8384]. Tempol decreases Lactobacillus, which has high BSH activity, and increases T-β-MCA content, which antagonizes intestinal FXR signaling and increases bile acid synthesis. Antagonizing intestinal FXR signaling also decreases circulating ceramides and inhibits de novo lipogenesis and hepatic gluconeogenesis [8485]. Fecal transplant of cecum microbiota from HFD-fed Fxr−/− mice and wild type mice into germ-free mice caused obesity [86]. These experiments indicate that FXR signaling may contribute to increased adiposity by altering the gut microbiota. A recent study reported that the anti-diabetic effect of metformin involved the reduction of B. fragilis and BSH activity [87]. This results in increased taurine (T)-UDCA and glycine (G)-UDCA, which antagonize intestinal FXR and improve hyperglycemia in diabetic patients. On the other hand, the intestine-specific FXR agonist fexaramine promotes adipose tissue browning and insulin sensitivity in mice [8889]. Fexaramine increases the LCA-producing gut bacteria Acetatifactor and Bacteroides, with both 7α- and 7β-dehydroxylase activities, to convert CDCA and UDCA to LCA. LCA stimulates TGR5/GLP-1 signaling to improve hepatic metabolism, and adipose tissue browning and energy metabolism [89]. Another study reported that gut commensal Bacteroides acidifaciens improved obesity and insulin sensitivity by increasing GLP-1 and decreasing intestinal dipeptidyl peptidase-4 (DDP-4) in mice [90]. This effect is apparently mediated by TGR5. Pro-hormone convertase 1/3 (PC1/3) cleaves preproglucagon to GLP-1, which is degraded by DDP-4. GLP-1 receptor agonists have been used to treat T2DM [91] and DDP-4 inhibitors (gliptins) reduce glucagon and increase serum GLP-1 levels and insulin sensitivity [92]. In contrast, another study indicated that activation of FXR in both enteroendocrine secretin tumor cells (STC-1) and in mice decreased proglucagon gene expression and GLP-1 secretion by interfering with carbohydrate responsive element binding protein gene expression and inhibiting glycolysis [93]. Therefore, intestinal FXR signaling can either aggravate or alleviate diabetes and obesity. It appears that FXR agonists and antagonists reshape the gut microbiota to exert differential effects on diabetes and obesity.
INTERACTIONS BETWEEN BILE ACIDS AND THE GUT MICROBIOTA
- Metabolic and bariatric surgery (MBG), such as Roux-en-Y gastric bypass (RYGB) and vertical sleeve gastrectomy (VSG), is the most effective way to reduce weight in overly obese patients [94]. Many recent studies report rapidly improved insulin sensitivity and diabetes 1 or 2 weeks following MBG, prior to weight loss, suggesting that metabolic changes are involved in improving glycemic control after gastric bypass. Serum total and primary bile acids, GLP-1, and FGF19 levels increase after MBG [9596979899], though the underlying mechanism for diabetes remission after MBG is not clear. Increased fasting serum bile acids, especially conjugated bile acids, implies a role for the gut microbiota in improving glycemic control and serum lipid profile in patients after gastric bypass [98].
- The metabolic benefits of MBG may be mediated by FXR/FGF19 signaling and TGR5/GLP-1 signaling [100]. The increased serum bile acids and FGF19 in T2DM patients after RYGB may indicate a dysregulation of the CYP7A1-FGF19 negative feedback pathway [101]. In obese human patients, circulating FGF21 is paradoxically induced, indicating FGF21 resistance, and bariatric surgery reduces serum FGF21 [102].
- In Fxr−/− and Tgr5−/− mice, the effect of VSG on glucose tolerance was reduced [103104]. Biliary diversion to the ileum also resulted in metabolic effects similar to RYGB in Tgr5−/−, but not intestine-specific Fxr−/− mice, suggesting a role of intestinal FXR in bile acid- and GLP-1-mediated metabolic improvement following bariatric surgery [105]. This study also suggested that intestinal bile acids and A. muciniphila may mediate these metabolic changes. It has been reported that A. muciniphila improves metabolism, inflammation, and outcomes of calorie restriction in obese patients [106107]. MBG may alter the enterohepatic circulation of bile acids, the gut microbiota, circulating bile acid composition, and bile acid pool size to improve metabolism in diabetes and obesity.
METABOLIC AND BARIATRIC SURGERY IMPROVES OBESITY AND DIABETES
- Bile acids as therapeutic drugs
- Bile acids have been used directly to treat diabetes and obesity. Rectal taurocholate administration increased GLP-1 secretion from L-cells, insulin secretion from β-cells, and decreased serum glucose and food intake in diabetic patients [108]. TCA is converted to DCA, which activates intestinal FXR and TGR5 signaling to improve glucose and insulin tolerance. CDCA increased adipose tissue browning in humans [47], while UDCA was used to treat obese patients in a small cohort study of morbid obesity [109]. Short term UDCA administration stimulated bile acid synthesis and reduced circulation of FGF19 by inactivating FXR. Bile acid and cholesterol synthesis was enhanced, though serum and liver triglycerides were increased, and stearoyl-CoA was induced in white adipose tissues, generating less toxic monosaturated fatty acids. Metformin alters the gut microbiota to increase TUDCA and GUDCA, which antagonize intestinal FXR to improve hyperglycemia in diabetic patients [87].
- Targeting FXR
- Targeting the bile acid receptors FXR and TGR5 has therapeutic potential for treating metabolic liver diseases [21]. Obeticholic acid (OCA, 6α-ethyl-CDCA) is a semisynthetic bile acid that activates FXR with 30-fold greater efficacy than CDCA. OCA inhibits bile acid synthesis, improves liver function, and reduces liver inflammation in primary biliary cirrhosis [110]. OCA is effective in improving NASH scores in clinical trials and is a promising drug therapy for NASH [111]. OCA inhibition of bile acid synthesis also induced the gram-positive bacteria Firmicutes and alleviated NASH in humans [112].
- Targeting TGR5
- TGR5-selective agonists have been shown to improve glucose homeostasis and metabolic diseases [113114]. Activation of TGR5 by a semisynthetic bile acid, INT-777 (6α-ethyl-23(S)-methylcholic acid), reduces macrophage inflammation, lipid loading and atherosclerosis by inhibiting nuclear factor κB (NF-κB) and proinflammatory cytokine production in low-density lipoprotein (LDL) receptor deficient mice [115]. TGR5 reduces NF-κB activation of proinflammatory cytokine production and reduces inflammation [116]. The TGR5-selective agonist INT-777, and FXR and TGR5 dual agonist INT-767 (6α-ethyl-3α, 7α, 23-trihydroxy-24-nor-5β-cholan-23-sulfate, sodium salt) promotes adipose tissue browning in mice [50]. Activation of both FXR and TGR5 also promotes GLP-1 secretion, improves glucose and lipid metabolism and reverses hepatic steatosis, insulin resistance, and CVD [50117118119120]. However, activation of TGR5 stimulates gallbladder proliferation and deficiency of TGR5 protects against cholesterol gallstone disease in mice [48]. Still, the negative effect of TGR5 activation in gallbladder in humans has not been reported. Intestine-selective TGR5 agonists could be developed for treating inflammatory bowel disease and diabetes.
- Bile acid sequestrants
- Bile acid sequestrants bind bile acids in the intestine to prevent bile acid reabsorption, thus reducing the bile acid pool size. This results in reduced FGF19 and increased CYP7A1 gene transcription and hepatic bile acid synthesis [121]. Bile acid sequestrants may increase DCA in the colon to stimulate TGR5-mediated secretion of GLP-1 [122]. By increasing bile acid synthesis, bile acid sequestrants increase hepatic LDL-cholesterol uptake, reducing hypercholesterolemia, but increase serum triglycerides and cause dyslipidemia [123]. Cholestyramine and colestipol are classic bile acid sequestrants used to treat cholesterol gallstone disease and hypercholesterolemia in human patients, while colesevelam, a second-generation bile acid sequestrant, improves glycemic control in T2DM patients [121124125].
- Targeting FGF19 and FGF21
- Patients with metabolic syndrome and obesity have reduced circulating FGF19 levels but increased FGF21 [102]. FGF19 increases energy metabolism and metabolic rate, reduces weight and improves glucose tolerance and insulin sensitivity in diet-induced obese mice [126]. Engineered FGF19 without tumorigenic activity may be useful to treat diabetes and obesity [127128129]. Conversely, serum FGF21 is increased in obese, diabetic and NAFLD patients [130131]. FGF21 is a metabolic regulator that also affects nutrient preference in humans and may be used to treat metabolic diseases [64132133134]. DPP4 is increased in islets of T2DM patients and DDP4 inhibitor-based therapies have had moderate success in reducing glucose intolerance and insulin resistance [135], while GLP-1 receptor agonists have shown promise for the management of T2DM and reducing weight [136].
BILE ACID-BASED THERAPY FOR DIABETES
- Emerging research in bile acid metabolism in the last three decades has contributed to identifying bile acids as endogenous ligands for FXR and TGR5 which mediate glucose, lipid, and energy metabolism and maintain whole body metabolic homeostasis. Overnutrition, high fat and high calorie diets, sleep disruption, drugs, and alcohol reshape gut microbiome to alter bile acid homeostasis and lead to dyslipidemia, hyperglycemia, insulin resistance and pathogenesis of diabetes, obesity, and related liver and heart diseases. Basic research in bile acid metabolism has contributed enormously to our current understanding of the molecular mechanisms and pathogenesis of liver-related metabolic diseases. However, most research has been focused on mouse models. Many of the results regarding FXR regulation of glucose, lipid, and energy metabolism are contradictory and remained to be resolved. Bile acid synthesis pathways are remarkably similar between mouse and human, but bile acid composition is very different. Thus, results from mouse studies cannot be extrapolated to humans without verification, and it is difficult to study bile acid metabolism in human subjects. Many metabolomic and microbiomic studies are limited to correlational analysis of serum and fecal samples between apparently healthy age- and sex-matched controls and patients diagnosed with diseases. Nevertheless, results from mouse studies have been translated to bile acid-based drugs targeting FXR, and to a lesser extent TGR5 for treating diabetes and obesity. Bile acid-based drugs are being developed for treating NASH, diabetes and obesity. However, bile acid-based FXR drugs cause pruritus and decrease serum high-density lipoprotein, and TGR5 drugs inhibit gallbladder emptying. Tissue selective FXR and TGR5 agonists and antagonists may be developed to circumvent the undesired side effects of drugs on other tissues. It is anticipated that bile acid-based drugs will be approved for treating diabetes, and treatments for NASH and fibrosis are expected in the near future.
CONCLUSIONS
-
Acknowledgements
- This work is supported by National Institutes of Health grants DK44442 and DK58379 to John Y. L. Chiang.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
NOTES
- 1. Rowley WR, Bezold C, Arikan Y, Byrne E, Krohe S. Diabetes 2030: insights from yesterday, today, and future trends. Popul Health Manag 2017;20:6-12. ArticlePubMedPMC
- 2. Finkelstein EA, Khavjou OA, Thompson H, Trogdon JG, Pan L, Sherry B, Dietz W. Obesity and severe obesity forecasts through 2030. Am J Prev Med 2012;42:563-570. ArticlePubMed
- 3. Targher G, Marchesini G, Byrne CD. Risk of type 2 diabetes in patients with non-alcoholic fatty liver disease: causal association or epiphenomenon? Diabetes Metab 2016;42:142-156. ArticlePubMed
- 4. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11-20. ArticlePubMedPDF
- 5. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J Hepatol 2018;68:335-352. ArticlePubMed
- 6. Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J Hepatol 2016;65:589-600. ArticlePubMed
- 7. Ma J, Hwang SJ, Pedley A, Massaro JM, Hoffmann U, Chung RT, Benjamin EJ, Levy D, Fox CS, Long MT. Bi-directional analysis between fatty liver and cardiovascular disease risk factors. J Hepatol 2017;66:390-397. ArticlePubMed
- 8. Chimakurthi CR, Rowe IA. Establishing the independence and clinical importance of non-alcoholic fatty liver disease as a risk factor for cardiovascular disease. J Hepatol 2016;65:1265-1266. ArticlePubMed
- 9. den Boer M, Voshol PJ, Kuipers F, Havekes LM, Romijn JA. Hepatic steatosis: a mediator of the metabolic syndrome. Lessons from animal models. Arterioscler Thromb Vasc Biol 2004;24:644-649. ArticlePubMed
- 10. Ginsberg HN, Zhang YL, Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obesity (Silver Spring) 2006;14(Suppl 1):41S-49S. ArticlePubMed
- 11. Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2010;7:195-203. ArticlePubMedPDF
- 12. Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 2016;150:1769-1777. ArticlePubMedPMC
- 13. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol 2016;11:451-496. ArticlePubMed
- 14. Haas JT, Francque S, Staels B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu Rev Physiol 2016;78:181-205. ArticlePubMed
- 15. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006;43(2 Suppl 1):S99-S112. ArticlePubMed
- 16. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 2009;50:1955-1966. ArticlePubMedPMC
- 17. Prawitt J, Caron S, Staels B. Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr Diab Rep 2011;11:160-166. ArticlePubMedPMCPDF
- 18. Haeusler RA, Astiarraga B, Camastra S, Accili D, Ferrannini E. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 2013;62:4184-4191. ArticlePubMedPMCPDF
- 19. Haeusler RA, Pratt-Hyatt M, Welch CL, Klaassen CD, Accili D. Impaired generation of 12-hydroxylated bile acids links hepatic insulin signaling with dyslipidemia. Cell Metab 2012;15:65-74. ArticlePubMed
- 20. Chiang JYL, Ferrell JM. Bile acid metabolism in liver pathobiology. Gene Expr 2018;18:71-87. ArticlePubMedPMC
- 21. Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res 2017;1:3-9. ArticlePubMedPMC
- 22. Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 1999;3:543-553. ArticlePubMed
- 23. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science 1999;284:1362-1365. ArticlePubMed
- 24. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999;284:1365-1368. ArticlePubMed
- 25. Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science 2002;296:1313-1316. ArticlePubMed
- 26. Goodwin B, Gauthier KC, Umetani M, Watson MA, Lochansky MI, Collins JL, Leitersdorf E, Mangelsdorf DJ, Kliewer SA, Repa JJ. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc Natl Acad Sci U S A 2003;100:223-228. ArticlePubMed
- 27. Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 2002;298:714-719. ArticlePubMed
- 28. Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, Dent P, Spiegel S, Shi R, Xu W, Liu X, Bohdan P, Zhang L, Zhou H, Hylemon PB. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012;55:267-276. ArticlePubMed
- 29. Raufman JP, Cheng K, Zimniak P. Activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig Dis Sci 2003;48:1431-1444. PubMed
- 30. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 2009;89:147-191. ArticlePubMed
- 31. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000;102:731-744. ArticlePubMed
- 32. Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 2014;66:948-983. ArticlePubMedPMC
- 33. Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, Kuipers F, Staels B. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes 2004;53:890-898. ArticlePubMedPDF
- 34. Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 2006;116:1102-1109. ArticlePubMedPMC
- 35. Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, Kuipers F, Staels B. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem 2006;281:11039-11049. ArticlePubMed
- 36. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, Edwards PA. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A 2006;103:1006-1011. ArticlePubMedPMC
- 37. Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, Mataki C, Sato H, Tanigawara Y, Schoonjans K, Itoh H, Auwerx J. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem 2011;286:26913-26920. ArticlePubMedPMC
- 38. Dufer M, Horth K, Wagner R, Schittenhelm B, Prowald S, Wagner TF, Oberwinkler J, Lukowski R, Gonzalez FJ, Krippeit-Drews P, Drews G. Bile acids acutely stimulate insulin secretion of mouse β-cells via farnesoid X receptor activation and K(ATP) channel inhibition. Diabetes 2012;61:1479-1489. ArticlePubMedPMCPDF
- 39. Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, Selbach O, Haussinger D, Kubitz R. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology 2007;45:695-704. ArticlePubMed
- 40. Keitel V, Ullmer C, Haussinger D. The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Biol Chem 2010;391:785-789. ArticlePubMed
- 41. Keitel V, Haussinger D. Role of TGR5 (GPBAR1) in liver disease. Semin Liver Dis 2018;38:333-339. ArticlePubMed
- 42. Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Haussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 2009;50:861-870. ArticlePubMed
- 43. Donepudi AC, Boehme S, Li F, Chiang JY. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology 2017;65:813-827. ArticlePubMedPDF
- 44. Alemi F, Poole DP, Chiu J, Schoonjans K, Cattaruzza F, Grider JR, Bunnett NW, Corvera CU. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 2013;144:145-154. ArticlePubMed
- 45. Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun 2008;372:78-84. ArticlePubMed
- 46. Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 2009;10:167-177. ArticlePubMedPMC
- 47. Broeders EP, Nascimento EB, Havekes B, Brans B, Roumans KH, Tailleux A, Schaart G, Kouach M, Charton J, Deprez B, Bouvy ND, Mottaghy F, Staels B, van Marken Lichtenbelt WD, Schrauwen P. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab 2015;22:418-426. ArticlePubMed
- 48. Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, Hoos L, Tetzloff G, Levitan D, Murgolo NJ, Keane K, Davis HR Jr, Hedrick J, Gustafson EL. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J 2006;398:423-430. ArticlePubMedPMCPDF
- 49. Vassileva G, Hu W, Hoos L, Tetzloff G, Yang S, Liu L, Kang L, Davis HR, Hedrick JA, Lan H, Kowalski T, Gustafson EL. Gender-dependent effect of Gpbar1 genetic deletion on the metabolic profiles of diet-induced obese mice. J Endocrinol 2010;205:225-232. ArticlePubMed
- 50. Pathak P, Liu H, Boehme S, Xie C, Krausz KW, Gonzalez F, Chiang JYL. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem 2017;292:11055-11069. ArticlePubMedPMC
- 51. Pathak P, Li T, Chiang JY. Retinoic acid-related orphan receptor α regulates diurnal rhythm and fasting induction of sterol 12α-hydroxylase in bile acid synthesis. J Biol Chem 2013;288:37154-37165. ArticlePubMedPMC
- 52. Ferrell JM, Chiang JY. Circadian rhythms in liver metabolism and disease. Acta Pharm Sin B 2015;5:113-122. ArticlePubMedPMC
- 53. Ferrell JM, Chiang JY. Short-term circadian disruption impairs bile acid and lipid homeostasis in mice. Cell Mol Gastroenterol Hepatol 2015;1:664-677. ArticlePubMedPMC
- 54. Donepudi AC, Ferrell JM, Boehme S, Choi HS, Chiang JYL. Deficiency of cholesterol 7α-hydroxylase in bile acid synthesis exacerbates alcohol-induced liver injury in mice. Hepatol Commun 2017;2:99-112. ArticlePubMedPMCPDF
- 55. Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, Ma J, Kemper B, Kemper JK. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014;516:108-111. ArticlePubMedPMCPDF
- 56. Wang Y, Ding Y, Li J, Chavan H, Matye D, Ni HM, Chiang JY, Krishnamurthy P, Ding WX, Li T. Targeting the enterohepatic bile acid signaling induces hepatic autophagy via a CYP7A1-AKT-mTOR axis in mice. Cell Mol Gastroenterol Hepatol 2016;3:245-260. ArticlePubMedPMC
- 57. Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, Goldstein JL, Brown MS. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci U S A 2012;109:16184-16189. ArticlePubMedPMC
- 58. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217-225. ArticlePubMed
- 59. Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009;49:297-305. ArticlePubMedPMC
- 60. Schlein C, Talukdar S, Heine M, Fischer AW, Krott LM, Nilsson SK, Brenner MB, Heeren J, Scheja L. FGF21 lowers plasma triglycerides by accelerating lipoprotein catabolism in white and brown adipose tissues. Cell Metab 2016;23:441-453. ArticlePubMed
- 61. Galman C, Lundasen T, Kharitonenkov A, Bina HA, Eriksson M, Hafstrom I, Dahlin M, Amark P, Angelin B, Rudling M. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab 2008;8:169-174. PubMed
- 62. Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, Burgess SC. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A 2009;106:10853-10858. PubMedPMC
- 63. Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, Chen JL, Jung DY, Zhang Z, Ko HJ, Kim JK, Veniant MM. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009;58:250-259. ArticlePubMedPMCPDF
- 64. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, Volk K, Kuo MS, Gordillo R, Kharitonenkov A, Scherer PE. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab 2013;17:790-797. ArticlePubMedPMC
- 65. Pathak P, Chiang JYL. Sterol 12α-hydroxylase aggravates dyslipidemia by activating the ceramide/mTORC1/SREBP1C pathway via FGF21 and FGF15. Gene Expr 2019 3 19 [Epub].Article
- 66. Li T, Chanda D, Zhang Y, Choi HS, Chiang JY. Glucose stimulates cholesterol 7alpha-hydroxylase gene transcription in human hepatocytes. J Lipid Res 2010;51:832-842. PubMedPMC
- 67. Li T, Francl JM, Boehme S, Ochoa A, Zhang Y, Klaassen CD, Erickson SK, Chiang JY. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem 2012;287:1861-1873. PubMed
- 68. Slatis K, Gafvels M, Kannisto K, Ovchinnikova O, Paulsson-Berne G, Parini P, Jiang ZY, Eggertsen G. Abolished synthesis of cholic acid reduces atherosclerotic development in apolipoprotein E knockout mice. J Lipid Res 2010;51:3289-3298. ArticlePubMedPMC
- 69. Kaur A, Patankar JV, de Haan W, Ruddle P, Wijesekara N, Groen AK, Verchere CB, Singaraja RR, Hayden MR. Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes 2015;64:1168-1179. ArticlePubMedPDF
- 70. Bertaggia E, Jensen KK, Castro-Perez J, Xu Y, Di Paolo G, Chan RB, Wang L, Haeusler RA. Cyp8b1 ablation prevents Western diet-induced weight gain and hepatic steatosis because of impaired fat absorption. Am J Physiol Endocrinol Metab 2017;313:E121-E133. ArticlePubMedPMC
- 71. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, Backhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013;17:225-235. ArticlePubMed
- 72. Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal Crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 2016;24:41-50. ArticlePubMed
- 73. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, Knight R. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 2018;15:397-411. ArticlePubMedPMCPDF
- 74. Ikegami T, Honda A. Reciprocal interactions between bile acids and gut microbiota in human liver diseases. Hepatol Res 2018;48:15-27. ArticlePubMedPDF
- 75. Patterson E, Ryan PM, Cryan JF, Dinan TG, Ross RP, Fitzgerald GF, Stanton C. Gut microbiota, obesity and diabetes. Postgrad Med J 2016;92:286-300. ArticlePubMedPDF
- 76. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009;58:1509-1517. ArticlePubMedPMCPDF
- 77. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55-60. ArticlePubMedPDF
- 78. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014;505:559-563. ArticlePubMedPDF
- 79. Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012;487:104-108. ArticlePubMedPMCPDF
- 80. Patankar JV, Wong CK, Morampudi V, Gibson WT, Vallance B, Ioannou GN, Hayden MR. Genetic ablation of Cyp8b1 preserves host metabolic function by repressing steatohepatitis and altering gut microbiota composition. Am J Physiol Endocrinol Metab 2018;314:E418-E432. ArticlePubMed
- 81. Chevre R, Trigueros-Motos L, Castano D, Chua T, Corliano M, Patankar JV, Sng L, Sim L, Juin TL, Carissimo G, Ng LFP, Yi CNJ, Eliathamby CC, Groen AK, Hayden MR, Singaraja RR. Therapeutic modulation of the bile acid pool by Cyp8b1 knockdown protects against nonalcoholic fatty liver disease in mice. FASEB J 2018;32:3792-3802. ArticlePubMedPDF
- 82. Gonzalez FJ, Jiang C, Patterson AD. An intestinal microbiota-farnesoid X receptor axis modulates metabolic disease. Gastroenterology 2016;151:845-859. ArticlePubMedPMC
- 83. Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, Tanaka N, Desai D, Amin SG, Albert I, Patterson AD, Gonzalez FJ. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 2015;125:386-402. ArticlePubMed
- 84. Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, Liu Y, Gavrilova O, Patterson AD, Gonzalez FJ. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 2015;6:10166ArticlePubMedPDF
- 85. Xie C, Jiang C, Shi J, Gao X, Sun D, Sun L, Wang T, Takahashi S, Anitha M, Krausz KW, Patterson AD, Gonzalez FJ. An intestinal farnesoid X receptor-ceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes 2017;66:613-626. ArticlePubMedPDF
- 86. Parseus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, Greiner TU, Perkins R, Backhed F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017;66:429-437. ArticlePubMed
- 87. Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, Liu J, Deng Y, Xia J, Chen B, Zhang S, Yun C, Lian G, Zhang X, Zhang H, Bisson WH, Shi J, Gao X, Ge P, Liu C, Krausz KW, Nichols RG, Cai J, Rimal B, Patterson AD, Wang X, Gonzalez FJ, Jiang C. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med 2018;24:1919-1929. ArticlePubMedPMCPDF
- 88. Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, Atkins AR, Khvat A, Schnabl B, Yu RT, Brenner DA, Coulter S, Liddle C, Schoonjans K, Olefsky JM, Saltiel AR, Downes M, Evans RM. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 2015;21:159-165. ArticlePubMedPMCPDF
- 89. Pathak P, Xie C, Nichols RG, Ferrell JM, Boehme S, Krausz KW, Patterson AD, Gonzalez FJ, Chiang JYL. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018;68:1574-1588. ArticlePubMedPMCPDF
- 90. Yang JY, Lee YS, Kim Y, Lee SH, Ryu S, Fukuda S, Hase K, Yang CS, Lim HS, Kim MS, Kim HM, Ahn SH, Kwon BE, Ko HJ, Kweon MN. Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal Immunol 2017;10:104-116. ArticlePubMedPDF
- 91. Nauck MA. Incretin-based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. Am J Med 2011;124(1 Suppl):S3-S18. ArticlePubMed
- 92. Ahren B, Landin-Olsson M, Jansson PA, Svensson M, Holmes D, Schweizer A. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J Clin Endocrinol Metab 2004;89:2078-2084. ArticlePubMed
- 93. Trabelsi MS, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, Perino A, Brighton CA, Sebti Y, Kluza J, Briand O, Dehondt H, Vallez E, Dorchies E, Baud G, Spinelli V, Hennuyer N, Caron S, Bantubungi K, Caiazzo R, Reimann F, Marchetti P, Lefebvre P, Backhed F, Gribble FM, Schoonjans K, Pattou F, Tailleux A, Staels B, Lestavel S. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat Commun 2015;6:7629ArticlePubMedPDF
- 94. Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Aminian A, Brethauer SA, Navaneethan SD, Singh RP, Pothier CE, Nissen SE, Kashyap SR. STAMPEDE Investigators. Bariatric surgery versus intensive medical therapy for diabetes: 5-year outcomes. N Engl J Med 2017;376:641-651. ArticlePubMedPMC
- 95. Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, Badman MK, Maratos-Flier E, Mun EC, Pihlajamaki J, Auwerx J, Goldfine AB. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 2009;17:1671-1677. ArticlePubMedPMCPDF
- 96. Pournaras DJ, Glicksman C, Vincent RP, Kuganolipava S, Alaghband-Zadeh J, Mahon D, Bekker JH, Ghatei MA, Bloom SR, Walters JR, Welbourn R, le Roux CW. The role of bile after Roux-en-Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology 2012;153:3613-3619. ArticlePubMedPMCPDF
- 97. Risstad H, Kristinsson JA, Fagerland MW, le Roux CW, Birkeland KI, Gulseth HL, Thorsby PM, Vincent RP, Engstrom M, Olbers T, Mala T. Bile acid profiles over 5 years after gastric bypass and duodenal switch: results from a randomized clinical trial. Surg Obes Relat Dis 2017;13:1544-1553. ArticlePubMed
- 98. Simonen M, Dali-Youcef N, Kaminska D, Venesmaa S, Kakela P, Paakkonen M, Hallikainen M, Kolehmainen M, Uusitupa M, Moilanen L, Laakso M, Gylling H, Patti ME, Auwerx J, Pihlajamaki J. Conjugated bile acids associate with altered rates of glucose and lipid oxidation after Roux-en-Y gastric bypass. Obes Surg 2012;22:1473-1480. ArticlePubMedPMCPDF
- 99. Nemati R, Lu J, Dokpuang D, Booth M, Plank LD, Murphy R. Increased bile acids and FGF19 after sleeve gastrectomy and Roux-en-Y gastric bypass correlate with improvement in type 2 diabetes in a randomized trial. Obes Surg 2018;28:2672-2686. ArticlePubMedPDF
- 100. Bozadjieva N, Heppner KM, Seeley RJ. Targeting FXR and FGF19 to treat metabolic diseases-lessons learned from bariatric surgery. Diabetes 2018;67:1720-1728. ArticlePubMedPMCPDF
- 101. Gerhard GS, Styer AM, Wood GC, Roesch SL, Petrick AT, Gabrielsen J, Strodel WE, Still CD, Argyropoulos G. A role for fibroblast growth factor 19 and bile acids in diabetes remission after Roux-en-Y gastric bypass. Diabetes Care 2013;36:1859-1864. ArticlePubMedPMCPDF
- 102. Gomez-Ambrosi J, Gallego-Escuredo JM, Catalan V, Rodriguez A, Domingo P, Moncada R, Valenti V, Salvador J, Giralt M, Villarroya F, Fruhbeck G. FGF19 and FGF21 serum concentrations in human obesity and type 2 diabetes behave differently after diet- or surgically-induced weight loss. Clin Nutr 2017;36:861-868. ArticlePubMed
- 103. Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Perez HE, Sandoval DA, Kohli R, Backhed F, Seeley RJ. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 2014;509:183-188. ArticlePubMedPMCPDF
- 104. McGavigan AK, Garibay D, Henseler ZM, Chen J, Bettaieb A, Haj FG, Ley RE, Chouinard ML, Cummings BP. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut 2017;66:226-234. ArticlePubMed
- 105. Albaugh VL, Banan B, Antoun J, Xiong Y, Guo Y, Ping J, Alikhan M, Clements BA, Abumrad NN, Flynn CR. Role of bile acids and GLP-1 in mediating the metabolic improvements of bariatric surgery. Gastroenterology 2019;156:1041-1051. ArticlePubMed
- 106. Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, Kayser BD, Levenez F, Chilloux J, Hoyles L. MICRO-Obes Consortium. Dumas ME, Rizkalla SW, Dore J, Cani PD, Clement K. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 2016;65:426-436. ArticlePubMed
- 107. Schneeberger M, Everard A, Gomez-Valades AG, Matamoros S, Ramirez S, Delzenne NM, Gomis R, Claret M, Cani PD. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci Rep 2015;5:16643ArticlePubMedPMCPDF
- 108. Adrian TE, Gariballa S, Parekh KA, Thomas SA, Saadi H, Al Kaabi J, Nagelkerke N, Gedulin B, Young AA. Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia 2012;55:2343-2347. ArticlePubMedPDF
- 109. Mueller M, Thorell A, Claudel T, Jha P, Koefeler H, Lackner C, Hoesel B, Fauler G, Stojakovic T, Einarsson C, Marschall HU, Trauner M. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol 2015;62:1398-1404. ArticlePubMedPMC
- 110. Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, Kowdley KV, Vincent C, Bodhenheimer HC Jr, Pares A, Trauner M, Marschall HU, Adorini L, Sciacca C, Beecher-Jones T, Castelloe E, Bohm O, Shapiro D. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015;148:751-761. ArticlePubMed
- 111. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E. NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015;385:956-965. ArticlePubMed
- 112. Friedman ES, Li Y, Shen TD, Jiang J, Chau L, Adorini L, Babakhani F, Edwards J, Shapiro D, Zhao C, Carr RM, Bittinger K, Li H, Wu GD. FXR-dependent modulation of the human small intestinal microbiome by the bile acid derivative obeticholic acid. Gastroenterology 2018;155:1741-1752. ArticlePubMedPMC
- 113. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov 2008;7:678-693. ArticlePubMedPDF
- 114. Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, Sardella R, Pruzanski M, Roda A, Pastorini E, Schoonjans K, Auwerx J. Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem 2009;52:7958-7961. ArticlePubMed
- 115. Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, Rizzo G, Gioiello A, Adorini L, Pellicciari R, Auwerx J, Schoonjans K. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab 2011;14:747-757. ArticlePubMedPMC
- 116. Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, Baldelli F, Donini A, Fiorucci S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS One 2011;6:e25637. ArticlePubMedPMC
- 117. Carino A, Cipriani S, Marchiano S, Biagioli M, Santorelli C, Donini A, Zampella A, Monti MC, Fiorucci S. BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci Rep 2017;7:42801ArticlePubMedPMCPDF
- 118. Zambad SP, Tuli D, Mathur A, Ghalsasi SA, Chaudhary AR, Deshpande S, Gupta RC, Chauthaiwale V, Dutt C. TRC210258, a novel TGR5 agonist, reduces glycemic and dyslipidemic cardiovascular risk in animal models of diabesity. Diabetes Metab Syndr Obes 2013;7:1-14. ArticlePubMedPMC
- 119. Miyazaki-Anzai S, Masuda M, Kohno S, Levi M, Shiozaki Y, Keenan AL, Miyazaki M. Simultaneous inhibition of FXR and TGR5 exacerbates atherosclerotic formation. J Lipid Res 2018;59:1709-1713. ArticlePubMedPMC
- 120. Ferrell JM, Pathak P, Boehme S, Gilliland T, Chiang JYL. Deficiency of both farnesoid X receptor and takeda g protein-coupled receptor 5 exacerbated liver fibrosis in mice. Hepatology 2019 1 21 [Epub].ArticlePDF
- 121. Smushkin G, Sathananthan M, Piccinini F, Dalla Man C, Law JH, Cobelli C, Zinsmeister AR, Rizza RA, Vella A. The effect of a bile acid sequestrant on glucose metabolism in subjects with type 2 diabetes. Diabetes 2013;62:1094-1101. ArticlePubMedPMCPDF
- 122. Potthoff MJ, Potts A, He T, Duarte JA, Taussig R, Mangelsdorf DJ, Kliewer SA, Burgess SC. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol 2013;304:G371-G380. ArticlePubMed
- 123. Herrema H, Meissner M, van Dijk TH, Brufau G, Boverhof R, Oosterveer MH, Reijngoud DJ, Muller M, Stellaard F, Groen AK, Kuipers F. Bile salt sequestration induces hepatic de novo lipogenesis through farnesoid X receptor- and liver X receptor alpha-controlled metabolic pathways in mice. Hepatology 2010;51:806-816. ArticlePubMed
- 124. Staels B, Kuipers F. Bile acid sequestrants and the treatment of type 2 diabetes mellitus. Drugs 2007;67:1383-1392. ArticlePubMed
- 125. Hansen M, Sonne DP, Mikkelsen KH, Gluud LL, Vilsboll T, Knop FK. Bile acid sequestrants for glycemic control in patients with type 2 diabetes: a systematic review with meta-analysis of randomized controlled trials. J Diabetes Complications 2017;31:918-927. ArticlePubMed
- 126. Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, Soriano R, Corpuz R, Moffat B, Vandlen R, Simmons L, Foster J, Stephan JP, Tsai SP, Stewart TA. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004;145:2594-2603. ArticlePubMed
- 127. Perry RJ, Lee S, Ma L, Zhang D, Schlessinger J, Shulman GI. FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Commun 2015;6:6980ArticlePubMedPDF
- 128. Degirolamo C, Sabba C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov 2016;15:51-69. ArticlePubMedPDF
- 129. Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun 2017;1:1024-1042. ArticlePubMedPMCPDF
- 130. Zhang X, Yeung DC, Karpisek M, Stejskal D, Zhou ZG, Liu F, Wong RL, Chow WS, Tso AW, Lam KS, Xu A. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 2008;57:1246-1253. ArticlePubMedPDF
- 131. Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, Ozdogan O, Imeryuz N, Kalayci C, Avsar E. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Invest 2010;40:887-892. ArticlePubMed
- 132. Kharitonenkov A, Shanafelt AB. FGF21: a novel prospect for the treatment of metabolic diseases. Curr Opin Investig Drugs 2009;10:359-364.PubMed
- 133. Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, Weng Y, Clark R, Lanba A, Owen BM, Brenner MB, Trimmer JK, Gropp KE, Chabot JR, Erion DM, Rolph TP, Goodwin B, Calle RA. A long-acting FGF21 molecule, PF-05231023, decreases body weight and improves lipid profile in non-human primates and type 2 diabetic subjects. Cell Metab 2016;23:427-440. ArticlePubMed
- 134. Talukdar S, Owen BM, Song P, Hernandez G, Zhang Y, Zhou Y, Scott WT, Paratala B, Turner T, Smith A, Bernardo B, Muller CP, Tang H, Mangelsdorf DJ, Goodwin B, Kliewer SA. FGF21 regulates sweet and alcohol preference. Cell Metab 2016;23:344-349. ArticlePubMed
- 135. Widlansky ME, Puppala VK, Suboc TM, Malik M, Branum A, Signorelli K, Wang J, Ying R, Tanner MJ, Tyagi S. Impact of DPP-4 inhibition on acute and chronic endothelial function in humans with type 2 diabetes on background metformin therapy. Vasc Med 2017;22:189-196. ArticlePubMedPMCPDF
- 136. Smits MM, van Raalte DH, Tonneijck L, Muskiet MH, Kramer MH, Cahen DL. GLP-1 based therapies: clinical implications for gastroenterologists. Gut 2016;65:702-711. ArticlePubMed
REFERENCES
Fig. 1
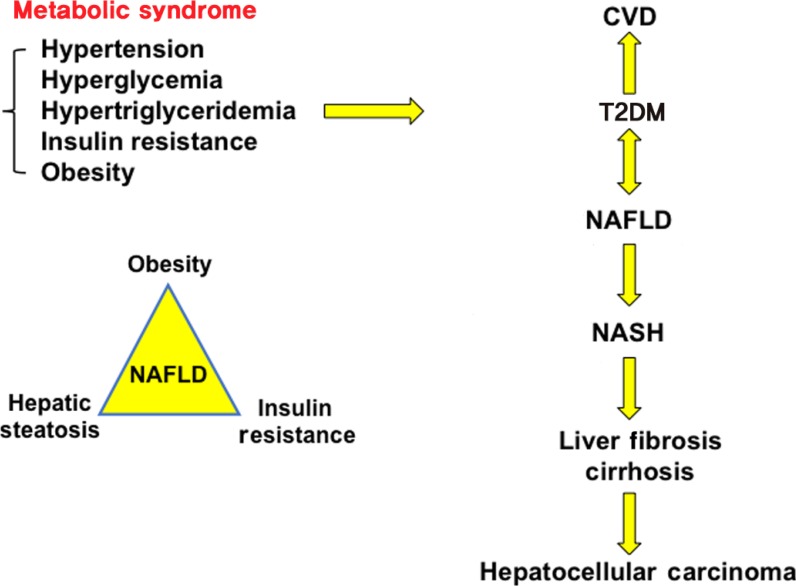
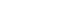
Metabolic syndrome is a collection of five phenotypes: hypertension, hyperglycemia, hypertriglyceridemia, insulin resistance and obesity. Many of these metabolic phenotypes are associated with type 2 diabetes mellitus (T2DM). T2DM increases risk for cardiovascular disease (CVD) and non-alcoholic fatty liver disease (NAFLD). NAFLD is a spectrum of liver disease including simple steatosis, nonalcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis, and hepatocellular carcinoma. Obesity, hepatic steatosis and insulin resistance all contribute to NAFLD.
Fig. 2
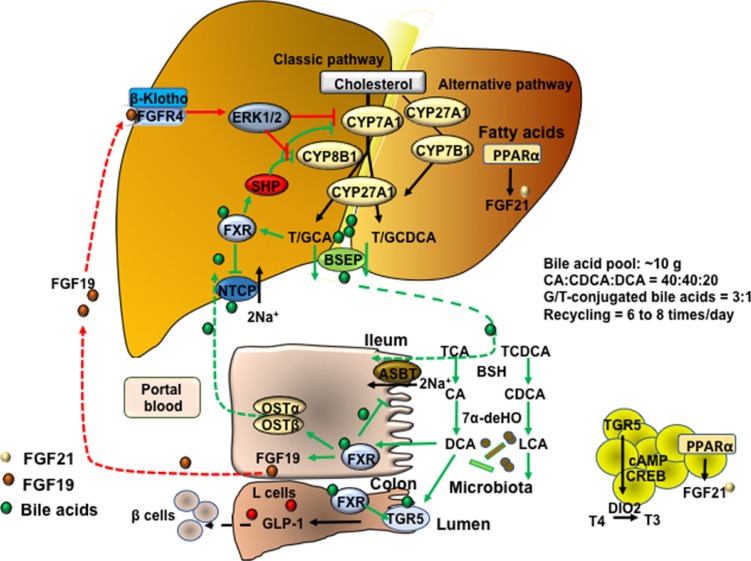
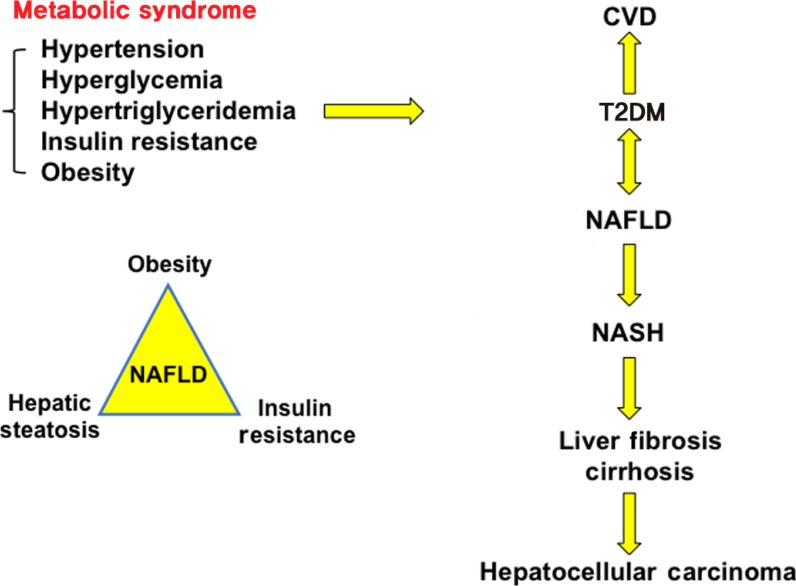
Bile acid synthesis, enterohepatic circulation of bile acids, and bile acid transport. In human hepatocytes, cholesterol 7α-hydroxylase (CYP7A1) catalyzes the first and rate-limiting step in the classic pathway of bile acid synthesis in which cholic acid (CA) and chenodeoxycholic acid (CDCA) are synthesized from cholesterol. Sterol 12α-hydroxylase (CYP8B1) is required for synthesis of CA, and without this enzyme CDCA is synthesized. The alternative pathway is initiated by sterol 27-hydroxylase (CYP27A1), which catalyzes steroid side-chain oxidation, followed by oxysterol 7α-hydroxylase (CYP7B1), which synthesizes the oxidized sterols that form CA and CDCA in hepatocytes. CYP7A1 is liver-specific, while CYP27A1 and CYP7B1 are expressed in extrahepatic tissues and macrophages. Bile acids are conjugated to the amino acids taurine (T) or glycine (G) for secretion into bile via bile salt export pump (BSEP). Bile acids are reabsorbed in the ileum via apical sodium-dependent bile acid transporter (ASBT) in enterocytes, where gut bacterial bile salt hydrolase (BSH) de-conjugates bile acids and 7α-dehydroxylase removes a hydroxyl group to form the secondary bile acids, deoxycholic acid (DCA) and lithocholic acid (LCA) from CA and CDCA, respectively. Bile acids are effluxed to portal blood via organic solute transporter α and β (OSTα/OSTβ) dimers and are transported to hepatocytes via Na2+-dependent taurocholate co-transporting peptide (NTCP) where they inhibit bile acid synthesis. Bile acids activate hepatic farnesoid X receptor (FXR) to induce small heterodimer partner (SHP), which inhibits CYP7A1 and CYP8B1 gene transcription. In enterocytes, bile acid activation of FXR induces fibroblast growth factor 19 (FGF19). FGF19 is transported to hepatocytes to activate FGF receptor 4 (FGFR4)/β-Klotho complex, which activates EKR1/2 signaling to inhibit CYP7A1 gene transcription. Bile acids activate Takeda G protein-coupled receptor 5 (TGR5) in intestinal L-cells, leading to secretion of glucagon-like peptide-1 (GLP-1), which stimulates insulin secretion from β-cells. In adipose tissue, activation of TGR5 stimulates cAMP/cAMP response element binding protein (CREBP) to induce thyroid hormone deiodinase type 2 (DIO2), which converts thyroxine (T4) to triiodothyronine (T3) and stimulates energy metabolism. ERK1/2, extracellular regulated kinase 1 and 2; PPARα, peroxisome proliferator-activated receptor α; GCA, glycocholic acid; GCDCA, glycochenodeoxycholic acid; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid.
Fig. 3

Nutrient regulation of bile acid synthesis, insulin signaling, and mechanistic target of rapamycin complex 1 (mTORC1) signaling. Feeding induces cholesterol 7α-hydroxylase (CYP7A1) but inhibits sterol 12α-hydroxylase (CYP8B1), while fasting inhibits CYP7A1 but induces CYP8B1. Feeding and fasting cycles affect bile acid synthesis and composition, which in turn regulate hepatic lipid and glucose metabolism. After feeding and during the postprandial state, bile acids are released from the gallbladder to aid in nutrient absorption. In hepatocytes, CYP7A1 and bile acid synthesis are stimulated to activate farnesoid X receptor (FXR) signaling and insulin/insulin receptor substrate 1 (IRS1)-AKT-phosphoinositide 3-kinase (PI3K) signaling. Insulin signaling inhibits mTORC1/protein S6 kinase (S6K) signaling and steroid regulatory element binding protein 1c (SREBP1c)-mediated lipogenesis. During the late post-prandial state, FXR induces fibroblast growth factor 19 (FGF19) to inhibit CYP7A1 and bile acid synthesis via FGF receptor 4 (FGFR4)/β-Klotho/extracellular regulated kinase 1 and 2 (ERK1/2) signaling. During fasting and prolonged starvation, free fatty acids released from adipose triglycerides activate peroxisome proliferator-activated receptor γ (PPARγ) in adipose tissue and PPARα in hepatocytes, and induce FGF21. FGF21 induces peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) to stimulate mitochondrial oxidative phosphorylation and energy production. FGF21 also inhibits mTORC1 signaling to stimulate insulin signaling. In enterocytes, FXR induces ceramides, which activate mTORC1/S6K signaling and stimulate processing of full length SREBP1c to its nuclear form (nSREBP1), stimulating lipogenesis. During fasting, CYP8B1 is induced and increases synthesis of cholic acid (CA) and deoxycholic acid (DCA). DCA activates intestinal FXR and ceramide synthesis. CYP8B1 inhibits FGF21 and activates mTORC1 signaling via inhibition of PPARα. CDCA, chenodeoxycholic acid; TCA, taurocholic acid; TGR5, Takeda G protein-coupled receptor 5; CREBP, cAMP response element binding protein; DIO2, deiodinase type 2; T, taurine; TCDCA, taurochenodeoxycholic acid; LCA, lithocholic acid; DCA, deoxycholic acid.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Gut microbiome-derived secondary bile acids: therapeutic targets for reducing cardiovascular disease in type 2 diabetes?
Sarah A. Johnson, Tiffany L. Weir
The American Journal of Clinical Nutrition.2024; 119(2): 241. CrossRef - Current updates on metabolites and its interlinked pathways as biomarkers for diabetic kidney disease: A systematic review
Soumik Das, V Devi Rajeswari, Ganesh Venkatraman, Ramprasad Elumalai, Sivaraman Dhanasekaran, Gnanasambandan Ramanathan
Translational Research.2024; 265: 71. CrossRef - Variant of the lactase LCT gene explains association between milk intake and incident type 2 diabetes
Kai Luo, Guo-Chong Chen, Yanbo Zhang, Jee-Young Moon, Jiaqian Xing, Brandilyn A. Peters, Mykhaylo Usyk, Zheng Wang, Gang Hu, Jun Li, Elizabeth Selvin, Casey M. Rebholz, Tao Wang, Carmen R. Isasi, Bing Yu, Rob Knight, Eric Boerwinkle, Robert D. Burk, Rober
Nature Metabolism.2024; 6(1): 169. CrossRef - Predictive value of serum TBA for 2-year MACEs in ACS patients undergoing PCI: a prospective cohort study
Wen Wen, Qinze Li, Jianqing She, Xiaofang Bai, Lisha Zhang, Ruifeng Li, Yan Wu, Juan Zhou, Zuyi Yuan
Scientific Reports.2024;[Epub] CrossRef - Collaborative Metabolism: Gut Microbes Play a Key Role in Canine and Feline Bile Acid Metabolism
John C. Rowe, Jenessa A. Winston
Veterinary Sciences.2024; 11(2): 94. CrossRef - The paradigm change from reactive medical services to 3PM in ischemic stroke: a holistic approach utilising tear fluid multi-omics, mitochondria as a vital biosensor and AI-based multi-professional data interpretation
Olga Golubnitschaja, Jiri Polivka, Pavel Potuznik, Martin Pesta, Ivana Stetkarova, Alena Mazurakova, Lenka Lackova, Peter Kubatka, Martina Kropp, Gabriele Thumann, Carl Erb, Holger Fröhlich, Wei Wang, Babak Baban, Marko Kapalla, Niva Shapira, Kneginja Ric
EPMA Journal.2024; 15(1): 1. CrossRef - Investigating the mechanism of cornel iridoid glycosides on type 2 diabetes mellitus using serum and urine metabolites in rats
Yadi Hou, Yanmei Huang, Zihui Shang, Shichao Ma, Tianyi Cui, Ali Chen, Yongxia Cui, Suiqing Chen
Journal of Ethnopharmacology.2024; 328: 118065. CrossRef - Improvement of myocardial injury and gut microbiota disturbance in type 2 diabetic mice by inulin with various degrees of polymerization
Siqiang Jia, Jianpeng Li, Bin Yu, Mengjie Li, Bo Cui
Food Bioscience.2023; 51: 102318. CrossRef - Bile acids and their receptors in regulation of gut health and diseases
Sen Lin, Sutian Wang, Peng Wang, Cuiming Tang, Zhenjiang Wang, Lian Chen, Guoqing Luo, Hong Chen, Yuntao Liu, Bin Feng, De Wu, Douglas G. Burrin, Zhengfeng Fang
Progress in Lipid Research.2023; 89: 101210. CrossRef - Effects of dietary oat supplementation on carcass traits, muscle metabolites, amino acid profiles, and its association with meat quality of Small-tail Han sheep
Li-wei Wang, Shao-feng Su, Jie Zhao, Xiao-long He, Shao-yin Fu, Biao Wang, Yun-fei Wang, Da-qing Wang, Na-na Yun, Xin Chen, Damien P Belobrajdic, Terigele, Xiao-dong Li, Li-li Jiang, Jiang-feng He, Yong-bin Liu
Food Chemistry.2023; 411: 135456. CrossRef - Noni (Morinda citrifolia L.) fruit polysaccharide ameliorated high-fat diet-induced obesity by modulating gut microbiota and improving bile acid metabolism
Wenjing Mo, Jiaqi Zou, Ming Wu, Zijun Peng, Wenjiang He, Wenzhi Li, Xiaoyong Wu
Journal of Functional Foods.2023; 101: 105408. CrossRef - Sodium glucose co-transporter 2 (SGLT2) inhibition via dapagliflozin improves diabetic kidney disease (DKD) over time associatied with increasing effect on the gut microbiota in db/db mice
Jiajia Wu, Yan Chen, Huinan Yang, Leyi Gu, Zhaohui Ni, Shan Mou, Jianxiao Shen, Xiajing Che
Frontiers in Endocrinology.2023;[Epub] CrossRef - Short communication: unique metabolic signature of proliferative retinopathy in the tear fluid of diabetic patients with comorbidities — preliminary data for PPPM validation
Martina Kropp, Eline De Clerck, Trong-Tin Kevin Steve Vo, Gabriele Thumann, Vincenzo Costigliola, Olga Golubnitschaja
EPMA Journal.2023; 14(1): 43. CrossRef - Impact of Vancomycin Treatment and Gut Microbiota on Bile Acid Metabolism and the Development of Non-Alcoholic Steatohepatitis in Mice
Kaichi Kasai, Naoya Igarashi, Yuki Tada, Koudai Kani, Shun Takano, Tsutomu Yanagibashi, Fumitake Usui-Kawanishi, Shiho Fujisaka, Shiro Watanabe, Mayuko Ichimura-Shimizu, Kiyoshi Takatsu, Kazuyuki Tobe, Koichi Tsuneyama, Yukihiro Furusawa, Yoshinori Nagai
International Journal of Molecular Sciences.2023; 24(4): 4050. CrossRef - Mitochondrial Cholesterol Metabolites in a Bile Acid Synthetic Pathway Drive Nonalcoholic Fatty Liver Disease: A Revised “Two-Hit” Hypothesis
Genta Kakiyama, Daniel Rodriguez-Agudo, William M. Pandak
Cells.2023; 12(10): 1434. CrossRef - Bile acid metabolism and signaling: Emerging pharmacological targets of dietary polyphenols
Kevin M. Tveter, Esther Mezhibovsky, Yue Wu, Diana E. Roopchand
Pharmacology & Therapeutics.2023; 248: 108457. CrossRef - Role of liver parameters in diabetes mellitus – a narrative review
Sana Rafaqat, Aqsa Sattar, Amber Khalid, Saira Rafaqat
Endocrine Regulations.2023; 57(1): 200. CrossRef - Bile acids induce IL-1α and drive NLRP3 inflammasome-independent production of IL-1β in murine dendritic cells
Ewa Oleszycka, Eoin C. O’Brien, Michael Freeley, Ed C. Lavelle, Aideen Long
Frontiers in Immunology.2023;[Epub] CrossRef - Machine learning for predicting diabetic metabolism in the Indian population using polar metabolomic and lipidomic features
Nikita Jain, Bhaumik Patel, Manjesh Hanawal, Anurag R. Lila, Saba Memon, Tushar Bandgar, Ashutosh Kumar
Metabolomics.2023;[Epub] CrossRef - The mitochondrial translocator protein (TSPO, 18 kDa): A key multifunctional molecule in liver diseases
Yuchang Li, Liting Chen, Vassilios Papadopoulos
Biochimie.2023;[Epub] CrossRef - Sodium+/taurocholate cotransporting polypeptide as target therapy for liver fibrosis
Ahmad Salhab, Johnny Amer, Yinying Lu, Rifaat Safadi
Gut.2022; 71(7): 1373. CrossRef - Comparative Evaluation of the Effect of Metformin and Insulin on Gut Microbiota and Metabolome Profiles of Type 2 Diabetic Rats Induced by the Combination of Streptozotocin and High-Fat Diet
Nan Hu, Qi Zhang, Hui Wang, Xuping Yang, Yan Jiang, Rong Chen, Liying Wang
Frontiers in Pharmacology.2022;[Epub] CrossRef - Immunomodulatory functions of FXR
Stefano Fiorucci, Angela Zampella, Patrizia Ricci, Eleonora Distrutti, Michele Biagioli
Molecular and Cellular Endocrinology.2022; 551: 111650. CrossRef - Production of New Microbially Conjugated Bile Acids by Human Gut Microbiota
Carlos J. Garcia, Vit Kosek, David Beltrán, Francisco A. Tomás-Barberán, Jana Hajslova
Biomolecules.2022; 12(5): 687. CrossRef - Regulation of the intestinal flora: A potential mechanism of natural medicines in the treatment of type 2 diabetes mellitus
liying he, Fang-Qing Yang, Pan Tang, Ting-Hui Gao, Cai-Xia Yang, Li Tan, Pan Yue, Ya-Nan Hua, Si-Jing Liu, Jin-Lin Guo
Biomedicine & Pharmacotherapy.2022; 151: 113091. CrossRef - Acrylamide induced glucose metabolism disorder in rats involves gut microbiota dysbiosis and changed bile acids metabolism
Zonghao Yue, Yanjuan Chen, Qian Dong, Dan Li, Meng Guo, Li Zhang, Yini Shi, Huiting Wu, Lili Li, Zhongke Sun
Food Research International.2022; 157: 111405. CrossRef - Effects of Herbal Therapy on Intestinal Microbiota and Serum Metabolomics in Different Rat Models of Mongolian Medicine
Guniang Jiu, Riao Dao, Dongxing Wu, Wang Hung, Haburi Jin, Li Li, Xiquan Fu, Chula Sa, Eerdunchaolu, Maulidiani .M
Evidence-Based Complementary and Alternative Medicine.2022; 2022: 1. CrossRef - The role of immune cells in the liver tumor microenvironment: an involvement of gut microbiota-derived factors
Tomonori Kamiya, Naoko Ohtani
International Immunology.2022; 34(9): 467. CrossRef - Reduced Cytokine Tumour Necrosis Factor by Pharmacological Intervention in a Preclinical Study
Armin Mooranian, Jacqueline Chester, Edan Johnston, Corina Mihaela Ionescu, Daniel Walker, Melissa Jones, Susbin Raj Wagle, Bozica Kovacevic, Thomas Foster, Momir Mikov, Hani Al-Salami
Biomolecules.2022; 12(7): 877. CrossRef - Crisis of the Asian gut: associations among diet, microbiota, and metabolic diseases
Phatthanaphong THERDTATHA, Akari SHINODA, Jiro NAKAYAMA
Bioscience of Microbiota, Food and Health.2022; 41(3): 83. CrossRef - Valorization of avocado seeds with antioxidant capacity using pressurized hot water extraction
Eng Shi Ong, Janelle Low, Joseph Choon Wee Tan, Su Yi Foo, Chen Huei Leo
Scientific Reports.2022;[Epub] CrossRef - The fungicide prothioconazole and its metabolite prothioconazole-desthio disturbed the liver-gut axis in mice
Lingyu Hu, Xiaofang Wang, Zhiwei Bao, Qihao Xu, Mingrong Qian, Yuanxiang Jin
Chemosphere.2022; 307: 136141. CrossRef - Gut Microbiota and Bile Acids Mediate the Clinical Benefits of YH1 in Male Patients with Type 2 Diabetes Mellitus: A Pilot Observational Study
Yueh-Hsiang Huang, Yi-Hong Wu, Hsiang-Yu Tang, Szu-Tah Chen, Chih-Ching Wang, Wan-Jing Ho, Yi-Hsuan Lin, Geng-Hao Liu, Pei-Yeh Lin, Chi-Jen Lo, Yuan-Ming Yeh, Mei-Ling Cheng
Pharmaceutics.2022; 14(9): 1857. CrossRef - Gut–Liver Axis and Non-Alcoholic Fatty Liver Disease: A Vicious Circle of Dysfunctions Orchestrated by the Gut Microbiome
Salvatore Pezzino, Maria Sofia, Gloria Faletra, Chiara Mazzone, Giorgia Litrico, Gaetano La Greca, Saverio Latteri
Biology.2022; 11(11): 1622. CrossRef - The Role of Bile Acids in Cardiovascular Diseases: from Mechanisms to Clinical Implications
Shuwen Zhang, Junteng Zhou, Wenchao Wu, Ye Zhu, Xiaojing Liu
Aging and disease.2022;[Epub] CrossRef - Role of bile acids in overweight and obese children and adolescents
Cosimo Giannini, Concetta Mastromauro, Serena Scapaticci, Cristina Gentile, Francesco Chiarelli
Frontiers in Endocrinology.2022;[Epub] CrossRef - Bile acids and microbes in metabolic disease
Dhiraj Kumar Sah, Archana Arjunan, Sun Young Park, Young Do Jung
World Journal of Gastroenterology.2022; 28(48): 6846. CrossRef - The Fungicide Prothioconazole and its Metabolite Prothioconazole-Desthio Disturbed the Liver-Gut Axis in Mice
Yuanxiang Jin, Lingyu Hu, Xiaofang Wang, Zhiwei Bao, Qihao Xu, Mingrong Qian
SSRN Electronic Journal .2022;[Epub] CrossRef - The Critical Effect of Bile Acids in Atherosclerosis
Shangwen Qi, Xu Luo, Shuangfang Liu, Bishi Ling, Hua Jin
Journal of Cardiovascular Pharmacology.2022; 80(4): 562. CrossRef - Role of sirtuin-1 (SIRT1) in hypoxic injury in pancreatic β-cells
Ye-Jee Lee, Esder Lee, Young-Hye You, Yu-Bae Ahn, Ki-Ho Song, Ji-Won Kim, Seung-Hyun Ko
Journal of Drug Targeting.2021; 29(1): 88. CrossRef - Impact of gut microbiota: How it could play roles beyond the digestive system on development of cardiovascular and renal diseases
Kanmani Suganya, Taekwon Son, Kyu-Won Kim, Byung-Soo Koo
Microbial Pathogenesis.2021; 152: 104583. CrossRef - Determination of bile acids from human gallbladder by 1H‐MRS—Protocol optimization and estimation of reproducibility
Peter Vermathen, Gaëlle Diserens, Dino Kröll, Philipp Nett, Guido Stirnimann, Reiner Wiest
NMR in Biomedicine.2021;[Epub] CrossRef - The gut microbiome-bile acid axis in hepatocarcinogenesis
Liwei Wu, Jiao Feng, Jingjing Li, Qiang Yu, Jie Ji, Jianye Wu, Weiqi Dai, Chuanyong Guo
Biomedicine & Pharmacotherapy.2021; 133: 111036. CrossRef - Gut microbiome and bile acids in obesity-related diseases
Rumei Li, Sergio Andreu-Sánchez, Folkert Kuipers, Jingyuan Fu
Best Practice & Research Clinical Endocrinology & Metabolism.2021; 35(3): 101493. CrossRef - Gut Microbiome of Indonesian Adults Associated with Obesity and Type 2 Diabetes: A Cross-Sectional Study in an Asian City, Yogyakarta
Phatthanaphong Therdtatha, Yayi Song, Masaru Tanaka, Mariyatun Mariyatun, Maisaroh Almunifah, Nancy Eka Putri Manurung, Siska Indriarsih, Yi Lu, Koji Nagata, Katsuya Fukami, Tetsuo Ikeda, Yuan-Kun Lee, Endang Sutriswati Rahayu, Jiro Nakayama
Microorganisms.2021; 9(5): 897. CrossRef - Microbial Ecosystem in Diabetes Mellitus: Consideration of the Gastrointestinal System
Awgichew Shewasinad Yehualashet, Berhan Begashaw Yikna
Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy.2021; Volume 14: 1841. CrossRef - Role of bile acids in inflammatory liver diseases
Ioannis Evangelakos, Joerg Heeren, Esther Verkade, Folkert Kuipers
Seminars in Immunopathology.2021; 43(4): 577. CrossRef - Plasma Bile Acid Profile in Patients with and without Type 2 Diabetes
Alessandro Mantovani, Andrea Dalbeni, Denise Peserico, Filippo Cattazzo, Michele Bevilacqua, Gian Luca Salvagno, Giuseppe Lippi, Giovanni Targher, Elisa Danese, Cristiano Fava
Metabolites.2021; 11(7): 453. CrossRef - Maternal cecal microbiota transfer rescues early-life antibiotic-induced enhancement of type 1 diabetes in mice
Xue-Song Zhang, Yue Sandra Yin, Jincheng Wang, Thomas Battaglia, Kimberly Krautkramer, Wei Vivian Li, Jackie Li, Mark Brown, Meifan Zhang, Michelle H. Badri, Abigail J.S. Armstrong, Christopher M. Strauch, Zeneng Wang, Ina Nemet, Nicole Altomare, Joseph C
Cell Host & Microbe.2021; 29(8): 1249. CrossRef - Pregnane X receptor exacerbates nonalcoholic fatty liver disease accompanied by obesity- and inflammation-prone gut microbiome signature
Sarah Kim, Sora Choi, Moumita Dutta, Jeffrey O. Asubonteng, Marianne Polunas, Michael Goedken, Frank J. Gonzalez, Julia Yue Cui, Maxwell A. Gyamfi
Biochemical Pharmacology.2021; 193: 114698. CrossRef - Gut microbiota as a target for prevention and treatment of type 2 diabetes: Mechanisms and dietary natural products
Fan Xia, Lu-Ping Wen, Bing-Chen Ge, Yu-Xin Li, Fang-Ping Li, Ben-Jie Zhou
World Journal of Diabetes.2021; 12(8): 1146. CrossRef - Association between circulating bile acid alterations and nonalcoholic steatohepatitis independent of obesity and diabetes mellitus
Youngae Jung, Bo Kyung Koo, Seo Young Jang, Dain Kim, Heeyeon Lee, Dong Hyeon Lee, Sae Kyung Joo, Yong Jin Jung, Jeong Hwan Park, Taekyeong Yoo, Murim Choi, Min Kyung Lee, Sang Won Kang, Mee Soo Chang, Won Kim, Geum‐Sook Hwang
Liver International.2021; 41(12): 2892. CrossRef - Bile acid activated receptors: Integrating immune and metabolic regulation in non-alcoholic fatty liver disease
Michele Biagioli, Stefano Fiorucci
Liver Research.2021; 5(3): 119. CrossRef - Synthesis of 12β-Methyl-18-nor-bile Acids
Andreas Luxenburger, Lawrence D. Harris, Elizabeth M. Ure, Roselis A. Landaeta Aponte, Anthony D. Woolhouse, Scott A. Cameron, Chris D. Ling, Ross O. Piltz, Andrew R. Lewis, Graeme J. Gainsford, Alex Weymouth-Wilson, Richard H. Furneaux
ACS Omega.2021; 6(38): 25019. CrossRef - Serum metabolomic biomarkers of perceptual speed in cognitively normal and mildly impaired subjects with fasting state stratification
Kamil Borkowski, Ameer Y. Taha, Theresa L. Pedersen, Philip L. De Jager, David A. Bennett, Matthias Arnold, Rima Kaddurah-Daouk, John W. Newman
Scientific Reports.2021;[Epub] CrossRef - Myocardial Infarction and Coronary Artery Disease in Menopausal Women With Type 2 Diabetes Mellitus Negatively Correlate With Total Serum Bile Acids
Xunxun Feng, Guangyao Zhai, Jiaqi Yang, Yang Liu, Yujie Zhou, Qianyun Guo
Frontiers in Endocrinology.2021;[Epub] CrossRef - An Integrated Bile Acids Profile Determination by UHPLC-MS/MS to Identify the Effect of Bile Acids Supplement in High Plant Protein Diet on Common Carp (Cyprinus carpio)
Xian Wei, Ting Yao, Fatou Ndoye Fall, Min Xue, Xiaofang Liang, Jie Wang, Wenlong Du, Xu Gu
Foods.2021; 10(10): 2465. CrossRef - Fatty liver index and development of cardiovascular disease in Koreans without pre-existing myocardial infarction and ischemic stroke: a large population-based study
Jun Hyung Kim, Jin Sil Moon, Seok Joon Byun, Jun Hyeok Lee, Dae Ryong Kang, Ki Chul Sung, Jang Young Kim, Ji Hye Huh
Cardiovascular Diabetology.2020;[Epub] CrossRef - Methionine restriction alleviates high-fat diet-induced obesity: Involvement of diurnal metabolism of lipids and bile acids
Luanfeng Wang, Bo Ren, Qian Zhang, Chuanqi Chu, Zhenting Zhao, Jianbin Wu, Weiyang Zhao, Zhigang Liu, Xuebo Liu
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease.2020; 1866(11): 165908. CrossRef - Bile Acids: Key Regulators and Novel Treatment Targets for Type 2 Diabetes
Yingjie Wu, An Zhou, Li Tang, Yuanyuan Lei, Bo Tang, Linjing Zhang
Journal of Diabetes Research.2020; 2020: 1. CrossRef - Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy
John Y. L. Chiang, Jessica M. Ferrell
American Journal of Physiology-Gastrointestinal and Liver Physiology.2020; 318(3): G554. CrossRef - Imidacloprid disturbed the gut barrier function and interfered with bile acids metabolism in mice
Guiling Yang, Xianling Yuan, Cuiyuan Jin, Dou Wang, Yanhua Wang, Wenyu Miao, Yuanxiang Jin
Environmental Pollution.2020; 266: 115290. CrossRef - Up to date on cholesterol 7 alpha-hydroxylase (CYP7A1) in bile acid synthesis
John Y.L. Chiang, Jessica M. Ferrell
Liver Research.2020; 4(2): 47. CrossRef -
Diabetes and Metabolism Journal in 2020: Good to Great
In-Kyung Jeong
Diabetes & Metabolism Journal.2020; 44(1): 1. CrossRef - Healthy dietary patterns to reduce obesity-related metabolic disease: polyphenol-microbiome interactions unifying health effects across geography
Camilla Diotallevi, Francesca Fava, Marco Gobbetti, Kieran Tuohy
Current Opinion in Clinical Nutrition & Metabolic Care.2020; 23(6): 437. CrossRef - Metabolic profiling of pre-gestational and gestational diabetes mellitus identifies novel predictors of pre-term delivery
Ilhame Diboun, Manjunath Ramanjaneya, Yasser Majeed, Lina Ahmed, Mohammed Bashir, Alexandra E. Butler, Abdul Badi Abou-Samra, Stephen L. Atkin, Nayef A. Mazloum, Mohamed A. Elrayess
Journal of Translational Medicine.2020;[Epub] CrossRef - Quantification of common and planar bile acids in tissues and cultured cells
Stephanie J. Shiffka, Jace W. Jones, Linhao Li, Ann M. Farese, Thomas J. MacVittie, Hongbing Wang, Peter W. Swaan, Maureen A. Kane
Journal of Lipid Research.2020; 61(11): 1524. CrossRef - Influência da microbiota intestinal na Doença Hepática Gordurosa Não Alcoólica
Isadora Barbosa de Almeida, Wermerson Assunção Barroso, Caroline Amélia Gonçalves
Revista Científica Multidisciplinar Núcleo do Conhecimento.2020; : 14. CrossRef - Ultra-Early and Early Changes in Bile Acids and Insulin after Sleeve Gastrectomy among Obese Patients
Adriana Florinela Cӑtoi, Alina Elena Pârvu, Aurel Mironiuc, Horațiu Silaghi, Ioana Delia Pop, Andra Diana Andreicuț
Medicina.2019; 55(12): 757. CrossRef
 PubReader
PubReader Cite
Cite